긴장성디스트로피와 뇌전증이 동반된 증례
Concurrence of Myotonic Dystrophy and Epilepsy
Article information
Myotonic dystrophy type 1 (DM1) is a clinically and genetically heterogeneous multisystem disorder characterized by progressive muscular dystrophy, myotonia, cardiac conduction defects, cataracts, and endocrine disorders [1]. Myotonic dystrophy associated with epilepsy is rare [2]. Here, we describe a patient with a temporal lobe epilepsy diagnosed with DM1 associated with medial temporal lesion.
Case
A 31-year-old man gradually developed gait disturbance and weakness in lower limbs. He often complained of stiffness and difficulty in releasing his grip following a handshake. He developed focal seizure in his teens. Cortical dysplasia or hippocampal sclerosis was suspected in brain magnetic resonance imaging (MRI) (Fig. 1). He was treated with anti-seizure medication at the time. The seizure did not recur afterward for several years and then the patient voluntarily discontinued the drug. Electroencephalogram showed normal awake patterns. Recently, there was no recurrence of seizures even without taking antiseizure medication. Neurological examination showed grip and percussion myotonia of the hand, tongue myotonia, proximal leg weakness, baldness, and temporalis atrophy. A motor power examination showed symmetrical proximal and distal muscle weakness (G4+/G4+) in whole limbs. Electromyography (EMG) revealed typical myotonic discharges on upper and lower limbs. In family history, there were no neuromuscular symptoms or signs in parents or younger brother at the time of genetic testing. Genetic testing of the DMPK gene on 19q13.2-q13.3 from peripheral blood leukocytes was performed using conventional polymerase chain reaction fragment analysis and repeat-primed polymerase chain reaction analysis. Genetic testing confirmed expansion in the DMPK gene with over 150 CTG repeats and a final diagnosis of DM1. Brain MRI (Fig. 1) at the time of 13 years old suggested right cortical dysplasia or hippocampal sclerosis with a few small high signal intensities in the right frontal white matter suggesting nonspecific microangiopathy. Otherwise, there was no other cortical dysplasia or developmental problems.
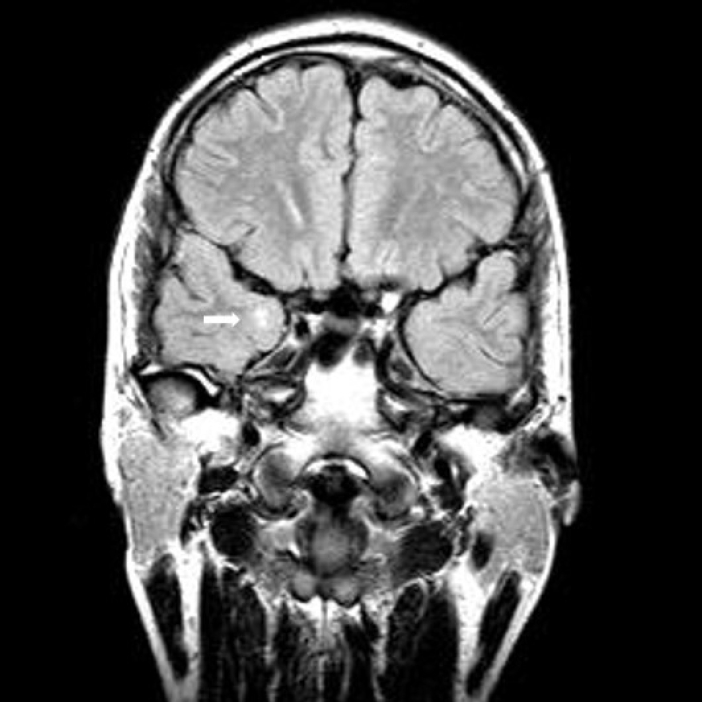
Coronal fluid attenuated inversion recovery image showing hyperintense lesion (arrow) in the medial subcortical part of right temporal lobe.
Discussion
Brain involvement is uncommon in DM1 [1,3]. It rarely occurs in hippocampal sclerosis. In affected persons, mental retardation is a frequent complication[1]. A patient treated with temporal lobe epilepsy for hippocampal sclerosis and later diagnosed with DM1 has not been reported yet. This case might very rare in terms of the disease itself. It might be delayed due to differences in interest in areas of neuromuscular disease specialty and epilepsy specialty that primarily see DM1. The diagnosis itself may be delayed because of the timing of the onset of symptoms. Therefore, it is crucial not only to investigate epilepsy onset in DM1, but also to know that DM1 might be the cause of temporal lobe epilepsy in patients with hippocampal sclerosis. Clinicians need to perform physical examination, EMG, and genetic study for this condition. An increase in diagnosis may extend to studies on the relationship between brain involvement and epilepsy in DM1, which is not yet known.