한국인 유년기 근위축측삭경화증 환자들의 임상적 특징
Clinical Characteristics of Korean Juvenile Amyotrophic Lateral Sclerosis
Article information
Trans Abstract
Background
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by progressive motor neuron degeneration with phenotypic heterogeneity, including age at onset. Juvenile ALS (JALS) includes ALS patients aged less than 25 years who typically show slow progression. This study aimed to identify the characteristics of juvenile ALS from Korean ALS cohorts.
Methods
We retrospectively investigated the clinical characteristics of JALS patients, who met the revised El Escorial-Airlie House criteria, in the Korean motor neuron disease cohort om January 2002 to November 2018. To evaluate the genetic background ofin JALS, we screened the SOD1 mutation in all JALS patients using PCR.
Results
Among the seven JALS patients, the mean age was 22.1 years (± 2.19 years) and 4 patients were male. Most patients were diagnosed within less than 12 months, but in one patient, it took 96 months to make the initial diagnosis. On assessing the cognitive function, none of the patients had dementia. The progression rate of JALS during follow-up was usually low (median [IQR], 0.31 [0.11-0.52]), except in patients with SOD1 mutation (3.40) and CLEC4C mutation (1.12). One patient revealed a family history of ALS with SOD1 mutation, but we also detected the SOD1 mutation among sporadic JALS patients.
Conclusions
Although JALS patients with genetic mutations (SOD1-p.Asn87Ser and CLEC4C-p.Lys210*) showed faster progression than other JALS patients, one patient with SOD1 mutation (p.Gly17Ala) showed slow progression. Familial ALS was rare; however, it might be caused by low or incomplete penetrance of the genes or by small number of JALS patients. To investigate the other genetic causes of JALS without the SOD1 mutation, a further study including detailed genetic analysis is needed.
서 론
근위축측삭경화증(amyotrophic lateral sclerosis, ALS)은 운동신경원세포의 퇴행성 변화로 인해 상위 운동신경원징후(upper motor neuron sign)와 하위 운동신경원징후(lower motor neuron sign)가 함께 나타나며, 지속적으로 진행하는 근 위축 및 근 위약을 보이는 대표적인 운동신경원병이다[1]. ALS는 다른 신경퇴행성 질환들과 마찬가지로 지역 및 국가, 인종에 상관없이 전 세계적으로 나이가 증가하면서 발병이 증가하는 양상을 보인다[2-7]. ALS의 임상 양상은 지역, 국가, 인종별로 차이를 보일 뿐 아니라, 같은 지역, 국가, 인종인 환자들 개개인 사이에서도 큰 차이를 보인다[8-12].
한국인을 대상으로 한 ALS의 역학 연구[2]에 따르면 일반적으로 증상 발현으로부터 38개월가량 경과 후 호흡부전으로 인해 사망에 이르게 되나, 증상 발생 나이에 따라 나누어 비교하였을 때 40세 이하에서 증상이 발병한 환자군의 생존 시간이 58개월로, 60세 이상 발병군의 생존시간(31개월)에 비해 훨씬 길게 보고되어 발병 연령의 차이가 예후의 차이를 보임을 확인하였다. 또한 국외의 연구들에 따르면, 25세 이하의 ALS 환자는 유년기 ALS (juvenile ALS)로 따로 분류하여 일반적인 ALS 환자에 비해 다른 임상적인 특징과 함께 양호한 예후를 보인다는 보고가 있다[12-17]. 국내에서도 병의 진행이 느린 유년기 ALS에 대한 3예의 증례보고가 있었으나, 이후 아직까지 국내에서는 유년기 ALS에 대한 자세한 조사는 이루어지지 않았다[18,19]. 이에 본 연구에서는 단일 상급 종합병원에서 추적관찰된 운동신경원병 코호트를 이용하여, 발병 연령이 25세 이하인 국내 유년기 ALS 환자들의 임상적 특징에 대해 조사하였다.
대상과 방법
1. 대상
본 연구는 2002년 1월 1일부터 2018년 11월 30일까지 한양대학교병원 운동신경원병 코호트에 등록된 환자 중, 발병 연령 25세 이하의 운동신경원병 환자를 대상으로 의무기록분석을 통해 후향적으로 조사하였다. 이 중 최초 방문 시 revised El Escorial criteria에 따라 clinically probable, definite ALS를 만족하는 ALS 환자와, revised El Escorial criteria에 따르면 clinically suspected 혹은 clinically possible ALS를 만족하지만 ALS의 가족력이 같이 확인된 가족성 ALS (familial ALS) 환자를 대상으로 하였다.
발병 연령 25세 이하의 운동신경원병 의심 환자 중 ALS가 아닌 다른 유사 질환으로 판명된 경우, 인종적 차이를 보여 이질적인 예후를 보일 것으로 예상되는 외국인 ALS 환자 및 임상정보가 불충분하여 진단을 내리기가 불가능한 경우 및 추적관찰이 불가능하여 예후를 알 수 없는 경우를 제외하였다.
최종적인 ALS의 진단 시점은 revised El Escorial criteria에 따라 clinically probable 혹은 definite ALS를 만족하며, 근전도 검사에서 운동신경원병을 시사하는 광범위한 탈분극 및 재분극 소견이 나오는 시점 혹은 clinically suspected 혹은 clinically possible ALS 환자 중 유전자 검사 혹은 가족력이 확인되는 시점을 기준으로 하였다.
2. 임상적 특징 조사
해당 환자들의 임상적 특징을 조사하기 위해, 조사 대상 환자의 성별, 발병 나이를 포함한 인구학적 특징, 증상의 첫 발생 시기, 증상의 첫 발현 부위(연수부위와 경추부 및 요추부), 처음 ALS로 진단받은 시기 및 처음 한양대학교병원 루게릭병 클리닉에 방문한 시기, 진단 당시의 기능적 손상(ALSFRS-R), 질환 진행속도(ΔFS) 및 첫 번째 방문 당시의 강제 폐활량(forced vital capacity, FVC)에 대해 조사하였다.
진단 당시 대상 환자의 질환 상태를 위하여 한글판 ALSFRS-R을 사용하여 12 항목에 대하여 각각 0점에서 4점까지 평가하여 기능적 손상을 평가하였다[20]. 첫 번째 방문 당시의 질환의 진행속도(ΔFS)는 증상이 시작되는 시점의 ALSFRS-R의 값, 즉 48점에서 첫 번째 방문 시 측정한 ALFFRS-R 값을 뺀 수치를 증상 발생시점부터 진단시점까지의 기간(개월)으로 나눈 값으로, 증상 발생부터 처음 방문하였을 때까지의 환자의 진행속도를 나타내는 척도로 정의하였다.
또한, 인지기능 장애의 동반 여부를 알기 위해 신경심리학적 검사를 통하여 인지기능 상태를 평가하였다. 신경심리검사에서 집행기능 장애 혹은 언어기능 장애가 있거나 둘이 같이 있는 경우 정의되었고, 장애의 기준은 정상 대조군 대비 16%ile (1 표준편차) 이하로 정의되었다. 이러한 평가의 결과를 바탕으로, 2017년 개정된 ALS-frontotemporal spectrum disorder (ALS-FTSD) 진단기준에 따라 정의된 인지장애(ALSci, ALS with cognitive impairment; ALSbi, ALS with behavioural impairment, ALScbi, ALS with combined cognitive and behavioural impairment) 및 전두측두엽치매(frontotemporal dementia) 여부를 정의하였다[21].
1) SOD1 유전자에 대한 유전학적 검사
ALS의 원인 유전자 중 가장 흔한 SOD1 유전자에 대한 검사를 위하여, 유전학적 검사에 대한 환자의 동의 하에 말초 혈액을 채취하여 말초혈액의 백혈구에서 Wizard Genomic DNA Purification kit의 표준적인 방법을 이용하여 Genomic DNA를 추출하였다. 이후 SOD1 gene의 5개의 exon에 대해 5쌍의 oligonucleotide primers를 이용하여 PCR 증폭하였고, 반응 산물은 Big dye terminator Ready Reaction kit (Applied Biosystems, Foster City, CA, USA)를 이용하여 ABI Prism 3130xl Genetic Analyzer를 통해 분석하였다. 이를 통해 모든 유년기 ALS 환자에서 SOD1 유전자에 대해 검사하였다.
결 과
조사기간 내 코호트에 등록된 2,374명의 환자 중 25세 이하의 유년기 ALS 환자는 총 7명(0.3%)으로 모두 이번 연구 대상으로 포함되었다. 모든 유년기 ALS 환자들의 임상적 특징에 대해 Table 1에 정리하여 기술하였다.
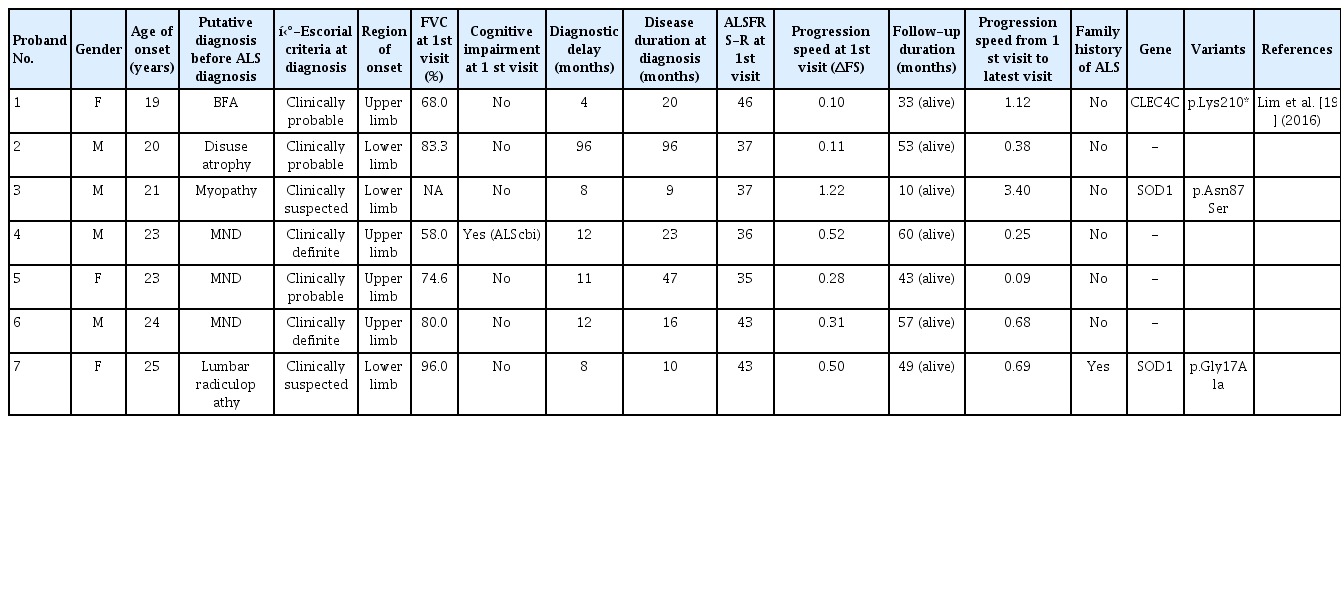
Clinical characteristics and prognosis of juvenile amyotrophic lateral sclerosis
총 7명의 유년기 ALS 환자 중, 5명의 환자들은 revised El-Escorial criteria에 의하면 clinically probable ALS 및 clinically definite ALS로 분류되어 진단되었다. 2명의 환자들(Proband 3, 7)에서는 상위 운동신경원징후가 발견되지 않아 clinically suspected ALS로 분류되었으나, SOD1 유전자 검사에서 기존에 보고된 유사 병원성 변이(likely pathogenic variant)가 확인되어 ALS로 진단되었다. 7명의 유년기 ALS 환자 중 6명에서는 확인된 가족력이 없어 산발형 ALS로 분류되었고, SOD1 유전자가 확인된 한 명의 환자(Proband 7)에서만 아버지가 ALS로 진단받고 사망한 병력이 있어 가족형 ALS로 분류되었다(Fig. 1). 또한 SOD1 유전자 이외의 유전자 변이는 1명의 환자(Proband 1)에서만 확인되었고, 이는 과거 2016년 보고에서 CLEC4C 유전자 변이가 발견된 환자이다[19]. 산발형 ALS 환자 중 유전자 변이가 발견된 환자들에 대해서는 가계도를 정리하였다(Fig. 2).
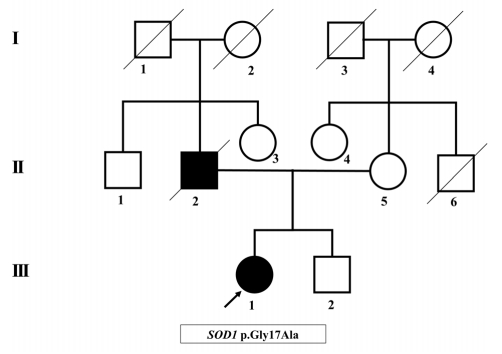
Pedigree of a case of familial amyotrophic lateral sclerosis (Proband 7). Squares indicate males; circles indicate females; filled symbols indicate affected individuals; diagonal lines across symbols indicate deceased individuals; arrows indicate the Probands.
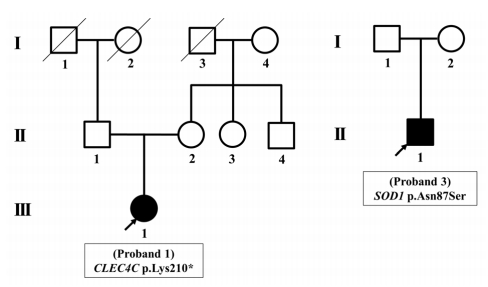
Pedigrees of sporadic amyotrophic lateral sclerosis patients who had genetic variants (Proband 1 and Proband 3). Squares indicate males; circles indicate females; filled symbols indicate affected individuals; diagonal lines across symbols indicate deceased individuals; arrows indicate the Probands.
모든 유년기 환자들이 처음 침범된 부위는 상지 혹은 하지로 확인되었고, 연수 부위의 발병은 없었다. 6명의 환자들에서 처음 방문 시의 FVC를 확인하였으며(median [IQR], 74.6 [63-88]%), 상지에서 처음 침범된 환자들(Proband 1, 4, 5, 6, median [IQR], 71.3 [60.5-78.7]%)이 하지에서 먼저 침범된 환자들(Proband 2,7, median 89.7%)에 비해 낮은 FVC를 보였다. 6명의 환자들은 발병 후 진단까지 걸리는 시간이 12개월 이하(median [IQR], 11 [8-12]개월)로 소요되었지만, 특징적으로 한 명의 환자(Proband 2)에서만 발병시기부터 확정진단까지 96개월의 매우 오랜 기간이 소요되었다. 첫 번째 방문 당시의 인지기능 장애에 대해 확인하였을 때, 2017년 개정된 ALS-FTSD 진단 기준에 의하면 한 명의 환자만 ALScbi로 진단되었지만, 전두측두엽 치매가 동반된 경우는 발견되지 않았다.
유년기 ALS 환자들이 한양대학교병원 루게릭병 클리닉에 첫 번째 내원하였을 때의 질환의 진행속도(ΔFS)를 확인하였을 때, 대부분의 환자에서는 질환의 진행속도(ΔFS)가 느리게 확인되었으나(median [IQR], 0.31 [0.11-0.52]), SOD1 유전자 변이가 확인된 한 명의 환자(Proband 3)에서만 ΔFS가 1.22로 첫 번째 내원시 빠른 진행을 보였다.
유년기 ALS 환자들은 코호트에 등록된 이후 최소 10개월 이상 병원에 내원하여 추적관찰(median [IQR], 49 [33-57]개월)하였다. 처음 방문시점부터 마지막 방문시점까지의 진행속도를 확인하였을 때, 발병 이후 처음 방문 시보다는 빠른 진행속도(median [IQR], 0.68 [0.25-1.12])를 보였다. 초기 내원 시 SOD1 유전자 변이와 함께 빠른 진행을 보였던 Proband 3의 진행속도는 3.40으로 더 빠르게 진행하는 양상을 보였으나, SOD1 유전자 변이가 확인된 다른 환자(Proband 7)은 0.68로 Proband 3에 비해 느린 진행속도를 보였다. 또한, 과거 2016년 보고에서 CLEC4C 유전자 변이가 발견된 Proband 1은 초기 내원까지는 느리게(0.10) 진행하였으나, 이후 다른 유년기 ALS 환자들에 비해 상대적으로 빠른 진행속도(1.12)를 보였다. 특히, SOD1의 변이를 가진 Proband 7은 마찬가지로 SOD1의 변이를 가지고 있는 Proband 3과 달리, 상대적으로 느린 진행을 보였다.
고 찰
유년기 ALS은 상위 및 하위 운동신경원을 모두 침범하며 진행하는 임상 양상을 가지는 운동신경원병 중 25세 이하의 발병 연령을 가지며, 전형적인 ALS과 다르게 매우 느린 진행을 보이는 것으로 알려져 있다[12-17].
본 연구에서 조사된 유년기 ALS 환자들의 내원 당시의 임상적 특징을 살펴볼 때, 여러 연구들에서 원인 유전자에 따라 연수부에서 발병하는 비율이 다르게 보고된데 비해[12-14], 본 연구에서는 연수부에서 발병한 경우는 확인되지 않았다. 하지만 이는 기존 40세 혹은 45세 이하에서 발병한다고 알려진 조기 발병형 ALS 환자가 일반적인 ALS 환자에 비해 연수부에서 발병하는 비율이 낮게 관찰된다고 알려진 바 있으며, 발병 연령 25세 이하의 유년기 ALS 환자도 조기 발병형 ALS 환자에 속해 이에 합당한 현상으로 생각된다[22-24]. 처음 방문 시 FVC를 확인하였을 때 상지에서 발병한 경우가 하지에서 발병한 경우보다 FVC가 낮게 나타나는 양상을 보였는데, 이는 일반적인 ALS에서 연수부를 제외한 척수부의 발병부위에 따른 FVC의 차이를 비교할 때 상하지에서의 차이가 없었던 과거 보고[25]와는 다른 결과로 확인되어 향후 유년기 ALS을 포함한 보다 많은 조기발병형 ALS 환자에서 추가적으로 확인해 볼 필요가 있다.
과거 연구에서 유년기 ALS에서는 인지기능 장애가 동반된 경우가 보고되지 않았으나[26], 본 연구에서는 FTD로 진단된 환자는 없었으나 한 명의 환자(Proband 4)에서 ALS cbi에 해당하는 인지기능 장애가 확인되었다. Proband 4의 인지기능 장애를 자세히 살펴볼 때, 집행기능(executive function)의 감소 및 탈억제(disinhibition)와 함께, 언어적 인출(retrieval) 장애, 시각적 재인변별력(recognition) 장애, 언어적 대면이름대기(confrontational naming) 장애를 보여 revised diagnostic criteria of ALS-FTSD에 따라 ALScbi로 정의되었으나, 해당 환자는 중국 국적의 조선족 교포로 한국어에 대한 이해가 완벽하지 않을 수 있어 한국인을 대상으로 한 검사 및 규준을 똑같이 적용하는 데 있어 의문점이 있다.
본 연구에서 발병 후 진단까지 비교적 빠른 시간(median [IQR], 11 [8-12]개월) 안에 진단되었지만, 특징적으로 Proband 2의 경우 96개월의 기간이 소요되었다. Proband 2는 진단 이전 6곳의 3차 종합병원을 내원하였으나 임상적으로 매우 느린 진행을 보이며, 근전도에서 ALS를 시사하는 광범위한 탈분극 및 재분극 소견이 보이지 않아 진단이 어려웠다. 처음 내원한 이후에도 상위 운동신경원 징후 및 하위 운동신경원 징후가 경추부 및 요추부의 2부위에서 같이 나오는 임상 양상을 보여 revised El Escorial criteria에 의해 clinically probable ALS에 해당하는 상태였으나, 내원 당시 상지에서는 근위부에만 근위축이 있으며 근전도 검사 에서는 근위부를 포함한 상하지 근육에서 광범위한 탈분극이 보이지 않아 운동신경원병에 합당하는 진단을 내리기 어려웠다. 하지만, 추적관찰을 시작한지 3개월 이후 연하장애 및 구음장애가 진행하며 7개월째 다시 검사한 근전도 검사에서 하지에서의 광범위한 탈분극이 확인되었고, 이후 상하지의 근력약화 및 근위축이 서서히 진행되며 12개월째 다시 검사한 근전도 검사에서 연수부 및 흉추부의 탈분극 소견을 포함한 하지의 광범위한 탈분극과 재분극 소견을 보여 최종적으로 유년기 ALS로 진단할 수 있었다. 이는 ALS의 조기 진단에 있어 근전도 검사를 통해 임상적으로 정상처럼 보이는 근육에서도 하위 운동신경원이 침범된 증거를 찾을수 있어 개정된 El Escorial criteria이 비해 근전도 검사를 이용해 하위 운동신경원 징후를 대체하는 Awaji criteria가 민감도가 더 높다고 여겨지는 일반적인 통념[27-29]에 반하는 경우로, 진행이 매우 늦은 환자에서 근위부가 먼저 침범한 경우 임상적으로 나타나는 하위 운동신경원 징후가 근전도 검사에서의 이상 소견보다 선행할 수 있는 가능성을 시사한다. 또한, 일반적인 ALS 환자에 비해 유년기 ALS에서 같은 부위 내(근위부에서 원위부 및 원위부에서 근위부) 혹은 다른 부위로의 파급(propagation)이 느리게 진행하는 것이 조기 발병형 ALS의 특징일 수 있어, 향후 이에 대해서는 추후 유년기 ALS 이외에도 일반적으로 느린 진행을 보인다고 알려진 조기 발병형 ALS 환자들에 대해서도 같은 현상들이 나타나는지에 대해 추가적인 연구가 필요할 것으로 보인다.
유년기 ALS 환자들에서는 상염색체 우성 유전 양상을 보이는 SOD1, FUS, SETX, TARDBP [12,17,30] 등의 유전자 뿐아니라, ALS2, SPG11, SIGMAR1 등의 상염색체 열성 유전양상을 보이는 유전자가 발견된 바 있어[31-33], 다양한 상염색체 연관 유전 양상을 보인다. 본 연구에서는 SOD1의 기존에 보고된 유사 병원성 변이(likely pathogenic variant)가 확인된 두 명의 환자들 중 한 명(Proband 7)에서만 가족력이 확인되어 가족형 ALS로 분류되었다. 기존 보고된 국내 ALS 환자에서의 유전자 연구[34]에 따르면, 가족형 ALS 환자의 비율은 3.5%이며, 그중 유전자 변이가 발견된 가족형 ALS 환자는 88.9%에 해당하지만, 이번 연구에서 가족력이 확인된 환자(Proband 7)에게서는 가족력이 있는 아버지가 환자의 진단 이전에 사망하여 부모의 유전자 검사를 시행할 수 없었고, 가족력이 없는 한 명의 환자(Proband 3)에서는 부모의 거절로 부모의 유전자 검사 및 자세한 가계도 조사를 시행하지 못하였다. 가족력이 없는 Proband 3의 경우, 부모에는 없이 새롭게 발견된 변이(de novo variant)일 수 있으나, 과거 SOD1 및 FUS 유전자를 동반한 경우에 불완전하거나 낮은 투과도(incomplete or low penetrance)를 보이는 경향이 있어 감퇴발현(reduced penetrance)한 가족형 ALS 환자와 산발형 ALS 환자를 구별하기 어렵다는 보고[17]가 있어, 부모에 대한 유전자 검사가 시행되지 않은 이상 부모에게서 유전되었을 가능성을 확인하기 어렵다.
본 연구에서 발병 후 진단까지의 기능적 장애 정도(ALSFRS-R) 및 질병의 진행속도(ΔFS)를 확인하였을 때, 기존에 알려진 대로 매우 느린 진행을 보이는 전형적인 유년기 ALS의 예후를 보이는 환자(Proband 2)와 함께 대부분의 환자들에서 느린 진행을 확인할 수 있었다. 하지만 SOD1 유전자의 유사 병원성 변이(p.Asn87Ser)가 발견된 한명의 환자(Proband 3)에서 다른 유년기 ALS 환자에 비해 매우 빠른 진행을 보이는 것이 확인되었다. 이에 대해 현재까지 보고된 문헌을 검색해본 바에 따르면, 일반적으로 유년기 ALS 환자가 느린 진행을 보이는데 비해 SOD1과 FUS 유전자가 발견된 유년기 ALS 환자들은 대부분 빠른 진행을 보이는 것으로 보고되어 있다[17]. 다만, SOD1 유전자의 유사 병원성 변이(p.Gly17Ala)가 발견된 다른 환자(Proband 7)에서는 Proband 3과 다르게 느린 진행을 보였는데, 해당 유전자 변이는 다른 보고[35]에서도 느린 진행을 보이는 것으로 알려져 있어 변이의 위치에 따른 다른 진행 양상을 보이는 것이 확인되었다. 또한, 과거 보고된 바 있는 CLEC4C 유전자 변이가 발견된 환자(Proband 1) 역시 초기 진단 당시에서는 느린 진행을 보였으나 이후 진행속도가 다른 환자들에 비해 빠른 진행(1.12)을 보였는데, plasmacytoid 수지상 세포(dendritic cells)에서 염증성 시토카인과 제1형 인터페론을 분비하는 데 관여하는 CLEC4C 유전자의 해당 변이(p.Lys210del)가 유년기 ALS의 질병 감수성 유전자(susceptibility gene)일 가능성을 시사한 기존 연구 논문에 가능성을 더하는 바가 있다[19]. 이로 미루어 일반적으로 알려진 바와 다르게 빠른 진행을 보이는 유년기 ALS 환자에서는 유전자 변이의 존재 여부에 대해 자세한 연구가 필요할 수 있다. 반면, 느리게 진행하는 유년기 ALS 환자에서도 다른 형태의 유전적 배경의 가능성이 있을 수 있어 이에 대한 추가적인 연구가 필요할 수 있다.
본 연구는 2004년 처음으로 국내에서 발표된 2예의 증례보고[18] 및 2016년에 발표된 CLEC4C 유전자의 변이가 발견된 1예의 보고[19] 이후 국내에서 2천 명 이상의 코호트 대상환자에서 조사된 유년기 ALS의 임상적 특징에 대한 연구로, 임상적으로 드문 유년기 ALS에 대해 7예의 보고와 함께 임상적 특징을 기술한 데에 의의가 있다. 하지만 기관절개 및 위루술 등의 구체적인 예후를 나타내는 종말점에 대한 조사가 부족한 부분이 있어 이번 연구에는 포함되지 못하였고, SOD1 이외의 ALS 연관 유전자에 대해 유전학적 분석을 시행하지 못한 한계가 있어, 추후 후속 연구를 통해 종말점까지의 예후가 아직까지 확인되지 않은 환자들에 대한 추가적인 예후 관찰 뿐 아니라, ALS와 연관된 유전자들에 대해 유전자 패널 혹은 차세대 염기서열 분석 등의 유전학적 분석을 통해 SOD1 이외의 다른 유전학적 특징에 대한 후속 연구를 시행해야 할 필요가 있다.