ņ×äņāüņĀüņ£╝ļĪ£ ņłśņØśņłśņČĢ ņØ┤ĒøäņŚÉ ĻĘ╝ņ£ĪņØś ņØ┤ņÖäņØ┤ ņ¦ĆņŚ░ļÉśļŖö ņ”ØņāüņØä ĻĘ╝ĻĖ┤ņןņ”Ø(myotonia)ņØ┤ļØ╝Ļ│Ā Ēæ£ĒśäĒĢśĻ│Ā ĻĘ╝ĻĖ┤ņןņ”ØņØ┤ ņ׳ņ£╝ļ®┤ ņ╣©ĻĘ╝ņĀäļÅäĻ▓Ćņé¼ļź╝ ĒåĄĒĢśņŚ¼ ĻĘ╝ĻĖ┤ņןļ░®ņĀäņØ┤ ļ│┤ņØ┤ļŖöņ¦Ć ĒÖĢņØĖĒĢ£ļŗż. ĻĘ╝ĻĖ┤ņןņ”ØņØĆ ĒŖ╣ņĀĢ ĻĘ╝ņ£Īņ¦łĒÖśņŚÉņä£ Ļ┤Ćņ░░ļÉśĻĖ░ ļĢīļ¼ĖņŚÉ ĻĘ╝ņ£Īņ¦łĒÖśņØś Ļ░Éļ│äņ¦äļŗ©ņŚÉ ļÅäņøĆņØ┤ ļÉśĻ│Ā, ņ╣©ĻĘ╝ņĀäļÅäĻ▓Ćņé¼ņŚÉņä£ ĻĘ╝ĻĖ┤ņןņĀäņ£äĻ░Ć ļ│┤ņØ┤ļ®┤ ĻĘ╝ĻĖ┤ņןļööņŖżĒŖĖļĪ£Ēö╝(myotonic dystrophy)ņÖĆ ļ╣äļööņŖżĒŖĖļĪ£Ēö╝ĻĘ╝ĻĖ┤ņןņ”ØņØä ņÜ░ņäĀ Ļ│ĀļĀżĒĢ┤ņĢ╝ ĒĢ£ļŗż. ņżæņŗ¼ĒĢĄĻĘ╝ņ£Īļ│æņ”Ø(centronuclear myopathy)ņØĆ ļō£ļ¼Ė ņäĀņ▓£ņä▒ ĻĘ╝ņ£Īļ│æņ”Øņ£╝ļĪ£ ņĀäĻĖ░ņ¦äļŗ©Ļ▓Ćņé¼ņŚÉņä£ ņ×Éļ░£ņĀäņ£äĻ░Ć ņŚåĻ│Ā ĻĘ╝ņ£Īļ│æņŚÉ ĒĢ®ļŗ╣ĒĢ£ ņÜ┤ļÅÖļŗ©ņ£ä ĒÖ£ļÅÖņĀäņ£äĻ░Ć ļ│┤ņØĖļŗż. ņĀĆņ×ÉļōżņØĆ ņ╣©ĻĘ╝ņĀäļÅäĻ▓Ćņé¼ņŚÉņä£ ļÜ£ļĀĘĒĢ£ ĻĘ╝ĻĖ┤ņןļ░®ņĀäņØä ļ│┤ņØĖ ņżæņŗ¼ĒĢĄĻĘ╝ņ£Īļ│æņ”Ø 1ņśłļź╝ ļ│┤Ļ│ĀĒĢśĻ│Āņ×É ĒĢ£ļŗż.
ņ”Ø ļĪĆ
30ņäĖ ļé©ņ×ÉĻ░Ć 10ļīĆ ņżæļ░śņŚÉ ļ░£ņāØĒĢ£ ļŗżļ”¼ņØś ĻĘ╝ļĀźņĀĆĒĢśļĪ£ ļé┤ņøÉĒĢśņśĆļŗż. ņ”ØņāüņØĆ ņä£ņä£Ē׳ ņ¦äĒ¢ēĒĢśņŚ¼ Ļ│äļŗ©ņśżļź┤ĻĖ░Ļ░Ć Ēלļōżņ¢┤ņ¦ĆĻ│Ā ĒīöņŚÉļÅä ĻĘ╝ļĀźņĀĆĒĢśĻ░Ć ļ░£ņāØĒĢśņśĆļŗż. Ļ░ÉĻ░ü ņ”Øņāü, ļ░£ņØīņØ┤ņāü, ĻĘ╝ņ£äņČĢņØĆ ļÅÖļ░śļÉśņ¦Ć ņĢŖņĢśļŗż. Ļ│╝Ļ▒░ļĀźĻ│╝ ņé¼ĒÜīļĀźņŚÉņä£ ĒŖ╣ņØ┤ ņé¼ĒĢŁņØ┤ ņŚåĻ│Ā ļ│ĄņÜ® ņżæņØĖ ņĢĮļ¼╝ļÅä ņŚåņŚłļŗż. Ļ░ĆņĪ▒ļĀźņŚÉņä£ ņÖĖĒĢĀļ©Ėļŗł, ņ¢┤ļ©Ėļŗł, ņØ┤ļ¬©, ņØ┤ņóģņé¼ņ┤īņŚÉņä£ ļ╣äņŖĘĒĢ£ ĻĘ╝ļĀźņĀĆĒĢśĻ░Ć ņ׳ņŚłļŗż.
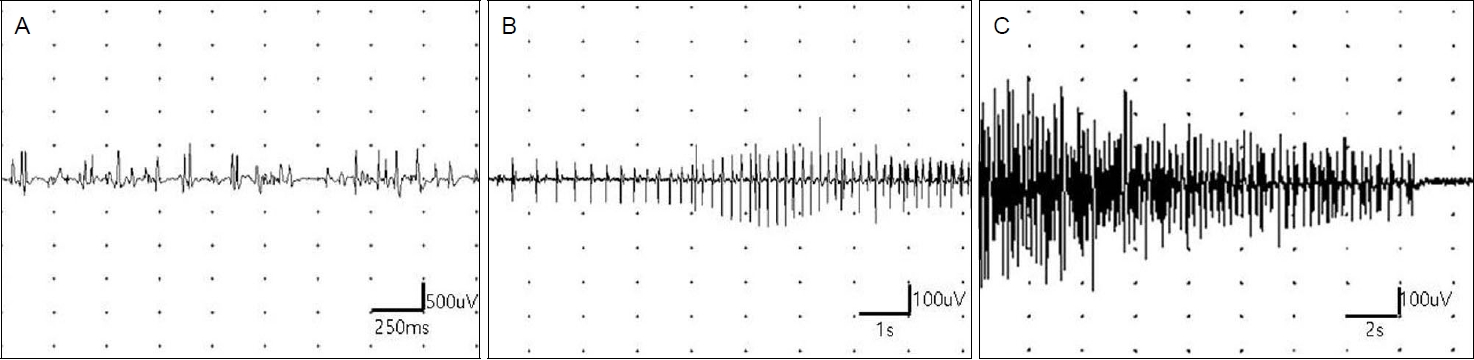
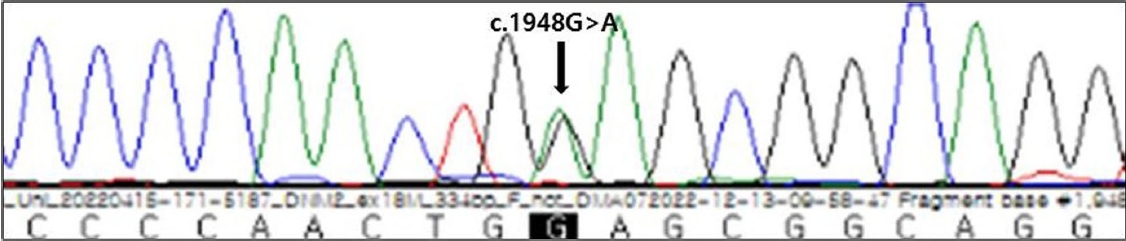
ņŗĀņ▓┤ņ¦äņ░░ņŚÉņä£ ņØ┤ļ¦łĒāłļ¬©Ļ░Ć ļ│┤ņśĆĻ│Ā. ņŗĀĻ▓ĮĻ│äņ¦äņ░░ņŚÉņä£ ņØśņŗØņØĆ ļ¬ģļŻīĒĢśņśĆĻ│Ā ļćīņŗĀĻ▓ĮĻĖ░ļŖźņØĆ Ļ▓ĮĒĢ£ ņĢłĻ▓ĆĒĢśņłśĻ░Ć ņ¢æņ¬ĮņŚÉņä£ ļ│┤ņØ┤ļŖö Ļ▓āņØä ņĀ£ņÖĖĒĢśĻ│ĀļŖö ņĀĢņāüņØ┤ņŚłļŗż. ĻĘ╝ļĀźņØĆ ņ¢æņ¬Į ĒĢśņ¦Ć ĻĘ╝ņ£äļČĆĻ░Ć Medical Research Council ļō▒ĻĖē 4+ņØ┤Ļ│Ā, ļéśļ©Ėņ¦ĆļŖö ņĀĢņāüņØ┤ņŚłņ£╝ļ®░ ĻĘ╝ņ£äņČĢņØ┤ļéś ĻĘ╝ļ╣äļīĆ ņåīĻ▓¼ņØĆ ņŚåņŚłļŗż. Ļ░ÉĻ░üĻ▓Ćņé¼ņÖĆ Ļ╣ŖņØĆĒלņżäļ░śņé¼ņŚÉņä£ ņĀĢņāüņØ┤ņŚłĻ│Ā ļ░öļ▒ģņŖżĒéż(babinski) ņ¦ĢĒøäļÅä ļ│┤ņØ┤ņ¦Ć ņĢŖņĢśļŗż. ĻĘ╝ņ£ĪņØś ņØ┤ņÖä ņ¦ĆņŚ░ņØ┤ļéś ĒāĆņ¦äĻĘ╝ĻĖ┤ņןņ”ØļÅä ļ│┤ņØ┤ņ¦Ć ņĢŖņĢśļŗż. ĒśłņĢĪĻ▓Ćņé¼ņŚÉņä£ Ēü¼ļĀłņĢäĒŗ┤ĒéżļéśņĀ£Ļ░Ć 552 IU/L (ņ░ĖĻ│Āņ╣ś <272 IU/L)ļĪ£ ņĢĮĻ░ä ņ”ØĻ░ĆĒĢśņśĆĻ│Ā ņĀäņ▓┤ĒśłĻĄ¼Ļ│äņé░Ļ▓Ćņé¼, ņĀäĒĢ┤ņ¦łĻ▓Ćņé¼, ņāØĒÖöĒĢÖĻ▓Ćņé¼, Ļ░æņāüņäĀĻĖ░ļŖźĻ▓Ćņé¼, ņĀüĒśłĻĄ¼ņ╣©Ļ░ĢņåŹļÅä, ņóģņ¢æĒæ£ņ¦Ćņ×É ļ░Å ņ×ÉĻ░Ćļ®┤ņŚŁņ¦łĒÖś Ļ┤ĆļĀ© ņØĖņ×ÉļōżņØĆ ņĀĢņāüņØ┤ņŚłļŗż. ņŗĀĻ▓ĮņĀäļÅäĻ▓Ćņé¼ļŖö ņĀĢņāüņØ┤ņŚłĻ│Ā, ĻĘ╝ņĀäļÅä Ļ▓Ćņé¼ņŚÉņä£ ĻĘ╝ņ£Īļ│æņØä ņŗ£ņé¼ĒĢśļŖö ņÜ┤ļÅÖļŗ©ņ£äĒÖ£ņä▒ņĀäņ£äĻ░Ć ļ│┤ņØ┤Ļ│Ā ļÜ£ļĀĘĒĢ£ ņĀäĻĖ░ņĀü ĻĘ╝ĻĖ┤ņןņ”ØņØ┤ ļ│┤ņśĆļŗż(Fig. 1). ņ░©ņäĖļīĆņŚ╝ĻĖ░ņä£ņŚ┤ļČäņäØ(next generation sequencing, NGS) ņ£ĀņĀäņ×ÉĻ▓Ćņé¼ņŚÉņä£ dynamin 2 (DNM2)ņØś ņ£ĀņĀäņ×Éļ│ĆņØ┤(c.1948G>A; p.Glu650Lys)Ļ░Ć Ļ┤Ćņ░░ļÉśņŚłļŗż(Fig. 2). ĻĘ╝ĻĖ┤ņןĻĘ╝ļööņŖżĒŖĖļĪ£Ēö╝ņÖĆ Ļ│Āņ╣╝ļź©ņŻ╝ĻĖ░ņä▒ļ¦łļ╣ä, ĒÅ╝ĒÄśļ│æņŚÉ ļīĆĒĢ£ dystrophia myotonica protein kinase (DMPK), sodium voltage gated channel alpha subunit 4 (SCNA4), alpha glucosidase (GAA) ņ£ĀņĀäņ×ÉļŖö ņØīņä▒ņ£╝ļĪ£ ļéśņÖöļŗż. ņ”ØļĪĆņØś ĒÖśņ×ÉļŖö ņāüņŚ╝ņāēņ▓┤ ņÜ░ņä▒ ņ£ĀņĀä, ĻĘ╝ņ£Īļ│æ ņ”ØņāüĻ│╝ ĻĘ╝ņĀäļÅä Ļ▓░Ļ│╝, ņ£ĀņĀäņ×É Ļ▓Ćņé¼ļź╝ ĒåĀļīĆļĪ£ DNM2ņ£ĀņĀäņ×Éļ│ĆņØ┤ ņżæņŗ¼ĒĢĄĻĘ╝ļ│æņ”Øņ£╝ļĪ£ ņ¦äļŗ©ĒĢśņśĆļŗż.
Ļ│Ā ņ░░
ņżæņŗ¼ĒĢĄĻĘ╝ņ£Īļ│æņ”ØņØĆ ļō£ļ¼Ė ņ£ĀņĀäņä▒ ĻĘ╝ņ£Īņ¦łĒÖśņ£╝ļĪ£ ņĪ░ņ¦üĒĢÖņĀüņ£╝ļĪ£ ĻĘ╝ņä¼ņ£Ā ņżæņĢÖņŚÉ ņ£äņ╣śĒĢ£ ĒĢĄĻ│╝ 1ĒśĢ ĻĘ╝ņä¼ņ£ĀņØś ņ£äņČĢņØ┤ļéś ņÜ░ņ£äĻ░Ć ĒŖ╣ņ¦ĢņØ┤ļŗż[1-3]. ņ£ĀņĀä ņ¢æņāüņŚÉ ļö░ļØ╝ XņŚ░Ļ┤Ć ņŚ┤ņä▒ĒśĢ, ļ│┤ĒåĄņŚ╝ņāēņ▓┤ ņÜ░ņä▒ĒśĢ, ļ│┤ĒåĄņŚ╝ņāēņ▓┤ ņŚ┤ņä▒ĒśĢņ£╝ļĪ£ ļéśļłäĻ│Ā, ņżæņ”ØļÅäļéś ņśłĒøäļÅä ņ£ĀņĀä ņ¢æņāüĻ│╝ Ļ┤ĆļĀ©ņØ┤ Ļ╣Ŗļŗż. XņŚ░Ļ┤Ć ņŚ┤ņä▒ĒśĢņØĆ ņČ£ņāØ ņŗ£ļČĆĒä░ ĻĘ╝ĻĖ┤ņןļÅäņĀĆĒĢś, ņĢłĻĄ¼ņÜ┤ļÅÖņןņĢĀ, ĒśĖĒØĪļČĆņĀäņØä ļ│┤ņØ┤Ļ│Ā, ļīĆļČĆļČä ņāØĒøä 1ļģä ļé┤ņŚÉ ņé¼ļ¦ØĒĢśļŖö Ļ░Ćņן ļéśņü£ ņśłĒøäļź╝ ļ│┤ņØĖļŗż. ļ│┤ĒåĄņŚ╝ņāēņ▓┤ ņŚ┤ņä▒ĒśĢņØĆ ļīĆļČĆļČä ņśüņ£ĀņĢäĻĖ░ņŚÉ ļ░£ļ│æĒĢśņŚ¼ ņä£ņä£Ē׳ ņ¦äĒ¢ēĒĢ£ļŗż. ļ│┤ĒåĄņŚ╝ņāēņ▓┤ ņÜ░ņä▒ĒśĢņØĆ ļīĆļČĆļČä ņ▓ŁņåīļģäĻĖ░ļéś ņä▒ņØĖĻĖ░ņŚÉ ļ░£ļ│æĒĢśĻ│Ā Ļ▓ĮĒĢ£ ĻĘ╝ņ£äļČĆ ĻĘ╝ļĀźņĀĆĒĢś, ņĢłĻ▓ĆĒĢśņłś, ņĢłĻĄ¼ņÜ┤ļÅÖņןņĢĀļź╝ ļ│┤ņØĖļŗż. ĒśłņĢĪĻ▓Ćņé¼ņŚÉņä£ Ēü¼ļĀłņĢäĒŗ┤ĒéżļéśņĀ£ļŖö ņĀĢņāüņØ┤Ļ▒░ļéś ņĢĮĻ░ä ņ”ØĻ░ĆļÉśņ¢┤ ņ׳ļŗż. ĻĄŁļé┤ņŚÉņä£ ļ│┤Ļ│ĀļÉ£ DNM2ņ£ĀņĀäņ×Éļ│ĆņØ┤ ņżæņŗ¼ĒĢĄĻĘ╝ļ│æņ”Ø ĒÖśņ×ÉļŖö ļ¬©ļæÉ ņåīņĢäņ▓ŁņåīļģäĻĖ░ņŚÉ ņé¼ņ¦ĆņØś ĻĘ╝ļĀźņĀĆĒĢśĻ░Ć ļ░£ņāØĒĢśņśĆĻ│Ā, ņĢłĻĄ¼ņÜ┤ļÅÖņןņĢĀĻ░Ć 75%ņŚÉņä£ ļÅÖļ░śļÉśņŚłĻ│Ā, ņĢłĻ▓ĆĒĢśņłś, ņĢłļ®┤ļ¦łļ╣ä, ņĢäĒé¼ļĀłņŖżĻ▒┤ ĻĄ¼ņČĢņ”Ø, ņ▓ÖņČöņĀäļ¦īņØĆ 50%ņŚÉņä£ ļÅÖļ░śļÉśņŚłļŗż[4]. ņ”ØļĪĆņØś ĒÖśņ×ÉļÅä ņ▓ŁņåīļģäĻĖ░ņŚÉ ĻĘ╝ļĀźņĀĆĒĢśĻ░Ć ļ░£ņāØĒĢśņśĆĻ│Ā ņĢłĻ▓ĆĒĢśņłśĻ░Ć ļÅÖļ░śļÉśņŚłņ£╝ļéś ļéśļ©Ėņ¦Ć ņ”ØņāüļōżņØĆ Ļ┤Ćņ░░ļÉśņ¦Ć ņĢŖņĢśļŗż.
ņĀäĒåĄņĀüņ£╝ļĪ£ ņĪ░ņ¦üĻ▓Ćņé¼ļĪ£ ņżæņŗ¼ĒĢĄĻĘ╝ņ£Īļ│æņ”ØņØä ņ¦äļŗ©ĒĢśņśĆņ¦Ćļ¦ī, ņżæņŗ¼ĒĢĄņØĆ ĻĘ╝ĻĖ┤ņןļööņŖżĒŖĖļĪ£Ēö╝, ļŗżņ¢æĒĢ£ ĻĘ╝ņ£Īļ│æņ”Ø, ļ¦īņä▒ ņŗĀĻ▓Įļ│æņ”Ø, ņ×¼ņāØ ĻĘ╝ņä¼ņ£ĀņŚÉņä£ļÅä Ļ┤Ćņ░░ļÉ£ļŗż[5]. ļ░śļ®┤ņŚÉ, ņ”ØņāüņØ┤ Ļ▓ĮĒĢ£ ņżæņŗ¼ĒĢĄĻĘ╝ņ£Īļ│æņ”ØņŚÉņä£ļŖö ņżæņŗ¼ĒĢĄņØ┤ Ļ┤Ćņ░░ļÉśņ¦Ć ņĢŖļŖö Ļ▓ĮņÜ░ļÅä ņ׳Ļ│Ā ņĪ░ņ¦üĻ▓Ćņé¼ļź╝ ņŗ£Ē¢ēĒĢ£ ĻĘ╝ņ£ĪņŚÉ ļö░ļØ╝ņä£ ņżæņŗ¼ĒĢĄņØś ļ╣łļÅäĻ░Ć ļŗ¼ļØ╝ņ¦äļŗż[6]. XņŚ░Ļ┤Ć ņŚ┤ņä▒ĒśĢņØĆ XņŚ╝ņāēņ▓┤ ņןņÖä(Xq28)ņŚÉ ņ£äņ╣śĒĢ£ myotubularin (MTM1) ņ£ĀņĀäņ×É, ļ│┤ĒåĄņŚ╝ņāēņ▓┤ ņŚ┤ņä▒ĒśĢņØĆ 2ļ▓łņŚ╝ņāēņ▓┤ ņןņÖä(2q14)ņŚÉ ņ£äņ╣śĒĢ£ amphiphysin 2 (BIN1) ņ£ĀņĀäņ×ÉņÖĆ 19ļ▓łņŚ╝ņāēņ▓┤ ņןņÖä(19q13.2)ņŚÉ ņ£äņ╣śĒĢ£ ryanodine receptor (RYR1)ņ£ĀņĀäņ×É, ļ│┤ĒåĄņŚ╝ņāēņ▓┤ ņÜ░ņä▒ĒśĢņØĆ 19ļ▓łņŚ╝ņāēņ▓┤ ļŗ©ņÖä(19p13.2)ņŚÉ ņ£äņ╣śĒĢ£ DNM2ņ£ĀņĀäņ×Éļ│ĆņØ┤ļĪ£ ņØĖĒĢ┤ ļ░£ņāØĒĢ£ļŗż[1,3]. ņĄ£ĻĘ╝ņŚÉ NGSļéś ņĀäņ▓┤ņ£ĀņĀäņ×ÉļČäņäØ(whole genome sequencing)ņØ┤ ļīĆļČĆļČäņØś ļ│æņøÉņŚÉņä£ Ļ░ĆļŖźĒĢ┤ņ¦Ćļ®┤ņä£ ņ£ĀņĀäņ×ÉĻ▓Ćņé¼Ļ░Ć ņĪ░ņ¦üĻ▓Ćņé¼ņØś ļČĆņĪ▒ĒĢ£ ņĀÉņØä ļ│┤ņÖä ļśÉļŖö ļīĆņ▓┤ĒĢśĻ│Ā ņ׳ļŗż. ĻĘ╝ņ£ĪņĪ░ņ¦üĻ▓Ćņé¼ ņĀäņŚÉ ņ£ĀņĀäņ×É Ļ▓Ćņé¼ļź╝ ņŗ£Ē¢ēĒĢśņŚ¼ ņ£ĀņĀäņä▒ ĻĘ╝ņ£Īņ¦łĒÖśņØä ņ£Āļ░£ĒĢśļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņ¦ä ņ£ĀņĀäņ×Éļ│ĆņØ┤Ļ░Ć ĒÖĢņØĖļÉśļ®┤ ņČöĻ░ĆņĀüņØĖ ņĪ░ņ¦üĻ▓Ćņé¼ļź╝ ņŗ£Ē¢ēĒĢśņ¦Ć ņĢŖĻĖ░ļÅä ĒĢ£ļŗż[2].
ņ╣©ĻĘ╝ņĀäļÅäĻ▓Ćņé¼ņŚÉņä£ ĻĘ╝ĻĖ┤ņןņĀäņ£äĻ░Ć ļ│┤ņØ┤ļŖö ļīĆĒæ£ņĀüņØĖ ņ¦łĒÖśņØĆ ĻĘ╝ĻĖ┤ņןļööņŖżĒŖĖļĪ£Ēö╝ņÖĆ ļ╣äļööņŖżĒŖĖļĪ£Ēö╝ĻĘ╝ĻĖ┤ņןņ”ØņØ┤Ļ│Ā ņØ┤ņÖĖņŚÉ Ļ│Āņ╣╝ļź©ņŻ╝ĻĖ░ņä▒ļ¦łļ╣ä, ļŗżļ░£ĻĘ╝ņ£ĪņŚ╝, Ļ░æņāüņäĀĻĖ░ļŖźņĀĆĒĢśņ”Ø, ĒÅ╝ĒÄśļ│æņŚÉņä£ļÅä Ļ░ĆļŖźĒĢśļŗż[7]. Ļ░Éļ│äņ¦äļŗ©ņØä ņ£äĒĢ┤ņä£ļŖö ņ×äņāüņĀü ĻĘ╝ĻĖ┤ņןņ”ØņØś ņ£Āļ¼┤Ļ░Ć ņżæņÜöĒĢśļ®░, ĻĘ╝ĻĖ┤ņןļööņŖżĒŖĖļĪ£Ēö╝ņÖĆ ļ╣äņŖżĒŖĖļĪ£Ēö╝ĻĘ╝ĻĖ┤ņןņ”Ø, Ļ│Āņ╣╝ļź©ņŻ╝ĻĖ░ņä▒ļ¦łļ╣äņŚÉņä£ļŖö ņ×äņāüņĀü ĻĘ╝ĻĖ┤ņןņ”ØņØ┤ ļÅÖļ░śļÉ£ļŗż. ĻĘ╝ĻĖ┤ņןļööņŖżĒŖĖļĪ£Ēö╝ļŖö ņ¦äĒ¢ēĒĢśļŖö ĻĘ╝ļĀźņĀĆĒĢśņÖĆ ļŗżņ¢æĒĢ£ ņĀäņŗĀ ņ”ØņāüņØ┤ ņ׳ļŗżļŖö ņĀÉņŚÉņä£ ļ╣äļööņŖżĒŖĖļĪ£Ēö╝ĻĘ╝ĻĖ┤ņןņ”Ø, Ļ│Āņ╣╝ļź©ņŻ╝ĻĖ░ņä▒ ļ¦łļ╣äņÖĆ Ļ░Éļ│äĒĢĀ ņłś ņ׳ļŗż. ņ”ØļĪĆņØś ĒÖśņ×ÉļŖö ņĀäĻĖ░ņĀü ĻĘ╝ĻĖ┤ņןņ”ØņØ┤ ļÜ£ļĀĘĒĢśņŚ¼ ņ×äņāüņĀü ĻĘ╝ĻĖ┤ņןņ”ØņØ┤ ņ׳ļŖöņ¦Ć ņŚ¼ļ¤¼ ņ░©ļĪĆ ĒÖĢņØĖĒĢśņśĆņ£╝ļéś ņØ┤ņÖäņ¦ĆņŚ░ņØ┤ļéś ĒāĆņ¦äĻĘ╝ĻĖ┤ņןņ”ØņØĆ ļ│┤ņØ┤ņ¦Ć ņĢŖņĢśļŗż. ĻĘ╝ĻĖ┤ņןņĀäņ£äĻ░Ć Ļ┤Ćņ░░ļÉśļŖö ņ¦łĒÖś ņżæņŚÉņä£ ĒÅ╝ĒÄśļ│æņØĆ ņ×äņāüņĀüņ£╝ļĪ£ ĻĘ╝ĻĖ┤ņןņ”Ø ņåīĻ▓¼ņØ┤ ļ│┤ņØ┤ņ¦Ć ņĢŖĻ│Ā ņ╣©ĻĘ╝ņĀäļÅäĻ▓Ćņé¼ņŚÉņä£ļ¦ī ĻĘ╝ĻĖ┤ņןņĀäņ£äĻ░Ć ļ│┤ņØĖļŗż. ņ”ØļĪĆņØś ĒÖśņ×ÉļŖö ĻĘ╝ĻĖ┤ņןļööņŖżĒŖĖļĪ£Ēö╝ņØś DMPKņ£ĀņĀäņ×ÉĻ▓Ćņé¼Ļ░Ć ņØīņä▒ņØ┤ņŚłļŗż. Ļ│Āņ╣╝ļź©ņŻ╝ĻĖ░ņä▒ļ¦łļ╣äņÖĆ ņäĀņ▓£ņØ┤ņāüĻĘ╝ĻĖ░ņןņ”Ø(paramyotnia congenita)ņØś SCN4Aļź╝ ĒżĒĢ©ĒĢ£ ņ£ĀņĀäņä▒ ĻĘ╝ņ£Īļ│æņ”ØņŚÉ ļīĆĒĢ£ NGSņŚÉņä£ DMN2ņ£ĀņĀäņ×Éļ│ĆņØ┤Ļ░Ć Ļ┤Ćņ░░ļÉśņŚłĻ│Ā ļéśļ©Ėņ¦ĆļŖö ļ¬©ļæÉ ņØīņä▒ņØ┤ņŚłļŗż. ļ│┤ĒåĄņŚ╝ņāēņ▓┤ ņŚ┤ņä▒ņ£ĀņĀäņØĖ ĒÅ╝ĒÄśļ│æĻ│╝ ņ£ĀņĀä ņ¢æņāüņØ┤ ļ¦×ņ¦Ć ņĢŖņ£╝ļéś ĻĘ╝ĻĖ┤ņןņ”Ø ņ”Øņāü ņŚåņØ┤ ĻĘ╝ĻĖ┤ņןņĀäņ£äļ¦ī ļ│┤ņŚ¼ņä£ ĒÅ╝ĒÄśļ│æņŚÉ ļīĆĒĢ£ ņ£ĀņĀäņ×ÉĻ▓Ćņé¼ļÅä ņŗ£Ē¢ēĒĢśņśĆĻ│Ā Ļ▓░Ļ│╝ļŖö ņØīņä▒ņØ┤ņŚłļŗż.
DNM2ļŖö GTPĻ▓░ĒĢ®Ļ░ĆņłśļČäĒĢ┤ĒÜ©ņåī(GTPase) ļŗ©ļ░▒ņ¦łņØä ļ░£ĒśäĒĢśļŖö ņ£ĀņĀäņ×ÉļĪ£ ņĢĪĒŗ┤ņäĖĒżĻ│©Ļ▓®(actin cytoskeleton) ņĪ░ļ”ĮĻ│╝ ņżæņŗ¼ņ▓┤(centrosome) Ļ▓░ĒĢ®ņŚÉ Ļ┤ĆņŚ¼ĒĢ£ļŗż[3]. ļśÉĒĢ£, DNM2ņ£ĀņĀäņ×Éļ│ĆņØ┤ ĒÖśņ×ÉņØś ņäĖĒżņŚÉņä£ Ēü┤ļØ╝ĒŖĖļ”░(clathrin) ļ¦żĻ░£ ņäĖĒżļé┤ ņØ┤ļÅÖņØś ņĀĆĒĢśļź╝ Ļ┤Ćņ░░ĒĢśņśĆĻ│Ā, ņØ┤ļŖö ļ¦ēņØ┤ļÅÖ(membrane trafficking)ņØś ņØ┤ņāüņØ┤ ļ│æĒā£ņāØļ”¼ņŚÉ Ļ┤ĆņŚ¼ĒĢ©ņØä ņŗ£ņé¼ĒĢ£ļŗż[8]. DNM2ņ£ĀņĀäņ×Éļ│ĆņØ┤ņŚÉ ņØśĒĢ£ ņ£ĀņĀäņä▒ ņŗĀĻ▓ĮĻĘ╝ņ£Īņ¦łĒÖśņØĆ ņĢäņŻ╝ ļō£ļ¼╝ņ¦Ćļ¦ī, ņżæņŗ¼ĒĢĄĻĘ╝ļ│æņ”ØņØä ņ£Āļ░£ĒĢśļŖö ņ£ĀņĀäņ×Éļ│ĆņØ┤ ņżæņŚÉņä£ļŖö ņĢĮ 50%ļź╝ ņ░©ņ¦ĆĒĢśļŖö Ļ░Ćņן ĒØöĒĢ£ ņ£ĀņĀäņ×ÉņØ┤ļŗż. ņżæņŗ¼ĒĢĄĻĘ╝ļ│æņ”ØņŚÉņä£ ĻĘ╝ĻĖ┤ņןņĀäņ£äĻ░Ć Ļ┤Ćņ░░ļÉ£ ņ”ØļĪĆĻ░Ć ļō£ļ¼╝Ļ▓ī ļ│┤Ļ│ĀļÉśņŚłĻ│Ā, ļ¬©ļæÉ DNM2ņ£ĀņĀäņ×Éļ│ĆņØ┤ņÖĆ Ļ┤ĆļĀ©ļÉśņ¢┤ ņ׳ņŚłļŗż[9,10]. ļśÉĒĢ£, ļ│Ė ņ”ØļĪĆņÖĆ ļ¦łņ░¼Ļ░Ćņ¦ĆļĪ£ ļ¬©ļōĀ ņ”ØļĪĆņŚÉņä£ ĻĘ╝ņĀäļÅäĻ▓Ćņé¼ņŚÉņä£ļ¦ī ĻĘ╝ĻĖ┤ņןņĀäņ£äĻ░Ć ļ│┤ņØ┤Ļ│Ā ĻĘ╝ĻĖ┤ņןņ”ØņØś ņ×äņāü ņ”ØņāüņØĆ ļ│┤ņØ┤ņ¦Ć ņĢŖņĢśļŗż.
ļ│Ė ņ”ØļĪĆņØś ĒÖśņ×ÉņÖĆ ņØ┤ņĀäņŚÉ ļ│┤Ļ│ĀļÉ£ ļ¼ĖĒŚīņØĆ ĻĘ╝ĻĖ┤ņןņĀäņ£äĻ░Ć DNM2ņ£ĀņĀäņ×Éļ│ĆņØ┤ ņżæņŗ¼ĒĢĄĻĘ╝ļ│æņ”ØņŚÉņä£ Ļ┤Ćņ░░ļÉĀ ņłś ņ׳ļŖö ĻĘ╝ņĀäļÅä ņåīĻ▓¼ņ×äņØä ļ│┤ņŚ¼ņżĆļŗż. ņ£ĀņĀäņä▒ ĻĘ╝ņ£Īļ│æņØ┤ ņØśņŗ¼ļÉśļŖö ĒÖśņ×ÉņŚÉņä£ ņĀäĻĖ░ņĀü ĻĘ╝ĻĖ┤ņןņ”ØņØ┤ ļ│┤ņØ┤ļ®┤ ņżæņŗ¼ĒĢĄĻĘ╝ļ│æņ”ØļÅä Ļ░Éļ│äņ¦äļŗ©ņŚÉ Ļ│ĀļĀżĒĢ┤ņĢ╝ ĒĢ£ļŗż.