Oculopharyngeal muscular dystrophy (OPMD) is a degenerative myopathy that mainly affects the ocular, bulbar and facial muscles [1]. The onset of the first symptom varies for people aged between 40 and 60 years, and all patients are symptomatic beyond the age of 70 [1,2]. The disease mainly presents ptosis and dysphagia, both of which show a slowly aggravating course. As the disease progresses, tongue atrophy, proximal limb weakness, and facial muscle weakness can occur [1].
Detection of the GCN trinucleotide expansions from 12 to 17 repeat in the first exon of polyadenylate-binding binding protein nuclear 1 (PABPN1) on chromosome 14q11 confirms the diagnosis [3]. Inheritance of OPMD is either an autosomal dominant or an autosomal recessive pattern depending on (GCN) size [2]. An autosomal dominant trait is caused by an expanded (GCN)12-17 repeat, producing an elongated polyalanine tract. Consequently, this domain forms stable, insoluble tubulofilamentous intranuclear inclusions (INIs) that are detected in skeletal muscles, being the hallmark of OPMD [4]. Homozygotes of (GCN)11 reveals an autosomal recessive form of OPMD [3]. Rarely, the heterozygotic combination of (GCN)12-17/(GCN)11 manifests relatively severe symptoms, probably due to the modifying effect of Ala11. There are 1-2% of (GCN)11 carriers in North American, European, and Japanese populations [2]. Hence, heterozygote (GCN)11 allele with normal allele has been considered as an insignificant mutation. However, a recent report suggests a plausible connection between heterozygous carrier of (GCN)10/(GCN)11 and clinical manifestation of OPMD by demonstrating the INIs in skeletal muscles [5,6].
Here, we report an additional case suggesting that the heterozygote (GCN)11 allele could present clinical manifestations of OPMD similar to the previous report.
Case
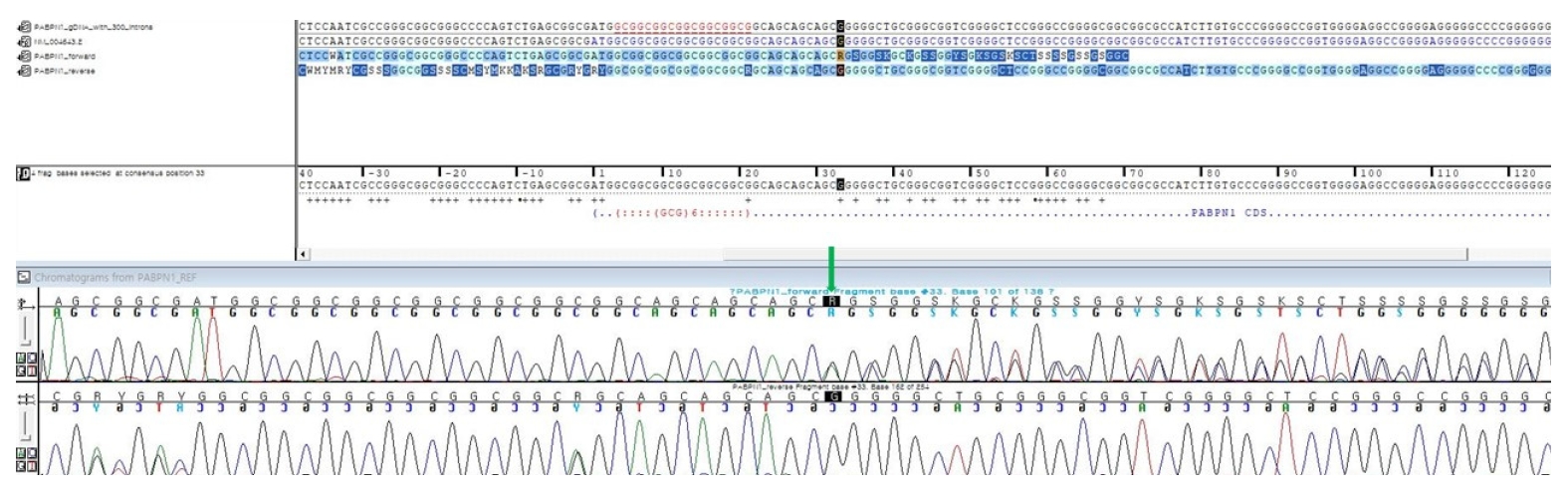
A 59-year-old female who had been suffering from dysarthria for 5 years visited the neurology department. She also complained about vague dysphagia. Symptoms did not worsen rapidly, only showing a mild defect. According to her medical history, she had hypertension and diabetes mellitus. Her parents died at 90 and 85 years old, without any neurologic symptoms. She had five siblings, and none of them displayed any evidence of neurodegenerative disease. Her neurological examination revealed dysarthria and dysphagia. She also showed an atrophied tongue and mild ptosis (Fig. 1). There was no diplopia. Limbs motor power was normal. Deep tendon reflexes and jaw jerk were normoactive. The evaluation focused on discovering diseases that can cause bulbar symptoms. The laboratory tests showed elevated fasting glucose and hemoglobin A1c that is compatible with diabetes mellitus. Creatine kinase level was 80 U/L. Brain magnetic resonance imaging and positron emission tomography-computed tomography were normal. An electrophysiological study showed that nerve conductions were normal. Electromyography revealed pathological spontaneous activities at rest (positive sharp waves and fasciculations) in bulbar muscles (masseter muscles). Motor unit potentials of bulbar muscles showed small amplitude (<0.3 mV) and shortened duration. The repetitive nerve stimulation test result was normal without any abnormal decrement or increment response. Considering her clinical history and neurological examination with electrophysiologic test results, we assumed this case as an atypical myopathy involving bulbar muscles. We checked acetylcholine receptor antibody titer to rule out myasthenia gravis and the titer was in the normal range. Muscle-specific tyrosine kinase antibody was negative. Next, we tested for the genetic defect causing myopathy. While other tests were normal, the PABPN1 gene test showed a heterozygous (GCG)6(GCA)4(GCG)/Ala11 expansion, another sequence presenting (GCN)10/Ala10 (Fig. 2). We concluded this case as an incomplete form of OPMD by single (GCN)11/Ala11 mutation. Unfortunately, an open skeletal muscle biopsy could not be performed.
Conclusion
When Brais and colleagues discovered the (GCN)11 allele, this variant only seemed to act as a modifier of a dominant phenotype or a recessive mutation [3,7]. However, a case from France confirmed that lone (GCN)11 could act as a dominant allele revealing an atypical clinical form of OPMD [5]. The patient had no known family history of OPMD. Dysphagia was the first symptom of the patient at the age of 67 years. He initially showed neither ptosis nor limitation in eye movements. Skeletal muscle biopsy revealed intranuclear inclusions consisted of tubular filaments [5]. Similar to the report mentioned above, our patient also had no family history of OPMD and showed symptoms limited to the bulbar region. Symptoms were slowly showing, which delayed the diagnosis. After additional workup and assessment, we reconsidered her disease as a mild form of OPMD that occurred by a single (GCN)11 expansion. In Korea, genetic confirmation of OPMD having (GCN)13/Ala13 variant has been reported previously. However, there is no additional information about the prevalence of OPMD or (GCN)11 carriers [8].
Unfortunately, our study had a few limitations. We could not obtain the genetic information of our subject’s parents or siblings. Therefore, whether this phenotype would be a result of de novo mutation is uncertain. Also, we could not acquire muscle tissue since the patient decided against an open skeletal muscle biopsy. However, we think that this report can support the hypothesis that single (GCN)11 with low penetrance can act dominantly, inducing symptoms compatible with OPMD [5]. As the polyalanine tract made by (GCN)11 is shorter than one by (GCN)12-17, a shorter tract can present mild problems such as lone bulbar symptom. Abnormal protein aggregation made by (GCN)17 directly impairs the function of the ubiquitin-proteasome pathway and the molecular chaperone, eventually inducing intranuclear inclusions [9]. We think similar pathology could be seen in (GCN)11 mutation revealing mild symptoms. Also, hidden modifier genes affecting the protein catabolism system can induce pathologic mechanisms among the carriers of (GCN)11. Since this hypothesis has been proven neither in vivo nor in vitro, further experimental studies would be needed. Investigating the frequency of the (GCN)11 allele in the general population and the prevalence of incomplete OPMD patients would be informative to understand the exact pathologic process of the disease.