ņä£ ļĪĀ
ņ£ĀņĀäĻ░Ģņ¦üĒĢśļ░śņŗĀļ¦łļ╣ä(hereditary spastic paraplegia)ļŖö ļŗ©ņØ╝ ņ£ĀņĀäņ×Éļ│ĆņØ┤ņŚÉ ņØśĒĢ£ ņŗĀĻ▓ĮĒć┤Ē¢ēņä▒ ņ¦łĒÖśņ£╝ļĪ£ ļ│ĆņØ┤ ņ£ĀņĀäņ×ÉņØś ņóģļźśņŚÉ ļö░ļØ╝ ļŗżņ¢æĒĢ£ ņ×äņāü ņ¢æņāüņØä ļ│┤ņØ╝ ņłś ņ׳ļŗż. ņĀä ņäĖĻ│äņĀüņØĖ ņ£Āļ│æļźĀņØĆ 10ļ¦ī ļ¬ģļŗ╣ 2-10ļ¬ģ ņĀĢļÅäļĪ£ ļ│┤Ļ│ĀļÉśĻ│Ā ņ׳ņ£╝ļéś[1,2], ĻĄŁļé┤ ĒÖśņ×ÉņØś ņ£Āļ│æļźĀņØĆ ņ▓┤Ļ│äņĀüņ£╝ļĪ£ ņĪ░ņé¼ļÉ£ ļ░öĻ░Ć ņŚåļŗż. ņ£ĀņĀäĻ░Ģņ¦üĒĢśļ░śņŗĀļ¦łļ╣äļŖö Ēö╝ņ¦łņ▓ÖņłśļĪ£ņØś Ēć┤Ē¢ēņä▒ ļ│ĆĒÖöļĪ£ ņØĖĒĢ£ ņ¦äĒ¢ēņä▒ĒĢśņ¦Ć Ļ░Ģņ¦üĻ│╝ ņŗ¼ļČĆĻ▒┤ļ░śņé¼ ĒĢŁņ¦äņØä ĒŖ╣ņ¦Ģņ£╝ļĪ£ ĒĢ£ļŗż. Ēśäņ×¼Ļ╣īņ¦Ć ņĢĮ 80ņŚ¼ Ļ░£ņØś ņøÉņØĖ(Ēøäļ│┤) ņ£ĀņĀäņ×ÉĻ░Ć ļ│┤Ļ│ĀļÉśņŚłņ£╝ļéś, ņ░©ņäĖļīĆ ņŚ╝ĻĖ░ņä£ņŚ┤ ļČäņäØ(next-generation sequencing, NGS) ļ░Å ņĀäņן ņŚæņå£ ņŚ╝ĻĖ░ņä£ņŚ┤ ļČäņäØ(whole exome sequencing) ĻĖ░ņłĀņØś ļ░£ņĀäņŚÉļÅä ļČłĻĄ¼ĒĢśĻ│Ā ņ£ĀņĀäĻ░Ģņ¦üĒĢśļ░śņŗĀļ¦łļ╣ä ņØśņŗ¼ ĒÖśņ×ÉņØś ņĀłļ░ś ņØ┤ņāüņŚÉņä£ ņ£ĀņĀäņ×É ļ│ĆņØ┤Ļ░Ć ĒÖĢņØĖļÉśņ¦Ć ņĢŖĻ│Ā ņ׳ļŗż[3-6].
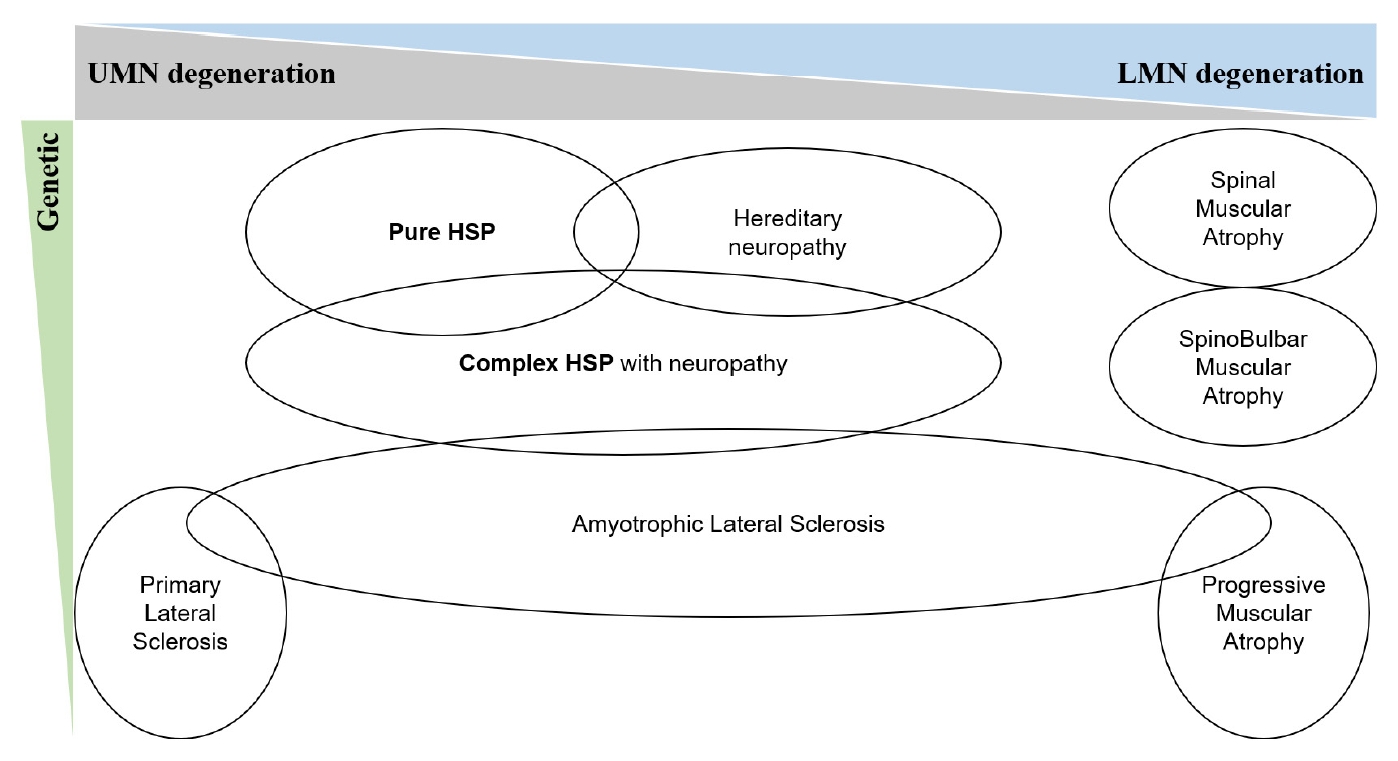
ņ£ĀņĀäĻ░Ģņ¦üĒĢśļ░śņŗĀļ¦łļ╣ä ĒÖśņ×ÉņŚÉņä£ ļÆżņä¼ņ£ĀĻĖ░ļæź(dorsal column) Ēć┤Ē¢ēņØ┤ ļÅÖļ░śļÉśļŖö Ļ▓ĮņÜ░Ļ░Ć ĒØöĒĢśĻ│Ā Ēö╝ņ¦łņ▓ÖņłśļĪ£ ņØ┤ņÖĖņŚÉļÅä ņČöņ▓┤ņÖĖļĪ£, ļ¦Éņ┤łņŗĀĻ▓Į ļō▒ ļŗżļźĖ ņŗĀĻ▓ĮĻ│äĻ░Ć ņ╣©ļ▓öļÉśĻĖ░ļÅä ĒĢśļéś, Ēü░ ļ▓öņŻ╝ņŚÉņä£ ņÜ┤ļÅÖņŗĀĻ▓ĮņäĖĒżļ│æņØś ĒĢ£ ņŖżĒÄÖĒŖĖļ¤╝ņ£╝ļĪ£ ļČäļźśĒĢĀ ņłś ņ׳ļŗż. ņÜ┤ļÅÖņŗĀĻ▓ĮņäĖĒżļ│æņØĆ ņ£äņÜ┤ļÅÖņŗĀĻ▓ĮņäĖĒż ņ╣©ļ▓ö ņŚ¼ļČĆņŚÉ ļö░ļØ╝ ņ£äņÜ┤ļÅÖņŗĀĻ▓ĮņäĖĒżļ│æĻ│╝ ņĢäļלņÜ┤ļÅÖņŗĀĻ▓ĮņäĖĒżļ│æņ£╝ļĪ£ ĻĄ¼ļČäĒĢĀ ņłś ņ׳ļŖöļŹ░, ņ£äņÜ┤ļÅÖņŗĀĻ▓ĮņäĖĒżļ│æņØś ļīĆĒæ£ņĀüņØĖ ĒśĢĒā£ņŚÉļŖö ņøÉļ░£ņä▒ņĖĪņéŁĻ▓ĮĒÖöņ”Ø(primary lateral sclerosis), ņ£ĀņĀäĻ░Ģņ¦üĒĢśļ░śņŗĀļ¦łļ╣ä ļō▒ņØ┤ ņ׳Ļ│Ā, ņĢäļלņÜ┤ļÅÖņŗĀĻ▓ĮņäĖĒżļ│æņØś ļīĆĒæ£ņĀüņØĖ ĒśĢĒā£ļĪ£ ņ▓ÖņłśĻĘ╝ņ£äņČĢņ”Ø(spinal muscular atrophy), ņ¦äĒ¢ēņä▒ĻĘ╝ņ£äņČĢņ”Ø(progressive muscular atrophy), ņøÉņ£äņ£ĀņĀäņÜ┤ļÅÖņŗĀĻ▓Įļ│æ(distal hereditary motor neuropathy) ļō▒ņØ┤ ņ׳ļŗż(Fig. 1). ņ£ĀņĀäĻ░Ģņ¦üĒĢśļ░śņŗĀļ¦łļ╣äļŖö ļ│ĆņØ┤ ņ£ĀņĀäņ×ÉņØś ņóģļźśņÖĆ ņĢäĒśĢņŚÉ ļö░ļØ╝ ļ¦żņÜ░ ļŗżņ¢æĒĢ£ ņ×äņāü ņ¢æņāüņØä ļ│┤ņØ╝ ņłś ņ׳ļŖö ņ¦łĒÖśĻĄ░ņ£╝ļĪ£ ļ│ĖĻ│ĀņŚÉņä£ļŖö ņ¦łļ│æņØś ļ¬©ļōĀ ņĖĪļ®┤ņØä ĒżĻ┤äņĀüņ£╝ļĪ£ ļŗżļŻ©ĻĖ░ļ│┤ļŗżļŖö ņŗżņĀ£ ņ¦äļŻī ĒśäņןņŚÉņä£ ļÅäņøĆņØ┤ ļÉĀ ņłś ņ׳ļŖö ņ¦äļŗ© ĒöäļĪ£ņäĖņŖżņÖĆ ņŻ╝ņÜö Ļ░Éļ│ä ņ¦äļŗ©ņŚÉ ļīĆĒĢ┤ ņĢīņĢäļ│┤Ļ│Āņ×É ĒĢ£ļŗż.
ļ│Ė ļĪĀ
1. ņ×äņāü ņ¢æņāü
ņ£ĀņĀäĻ░Ģņ¦üĒĢśļ░śņŗĀļ¦łļ╣äļŖö Ēö╝ņ¦łņ▓ÖņłśļĪ£ņØś Ēć┤Ē¢ēņä▒ ļ│ĆĒÖöņŚÉ ņØśĒĢ£ ņ¦äĒ¢ēņä▒ ĒĢśņ¦Ć Ļ░Ģņ¦üņØä ĒŖ╣ņ¦Ģņ£╝ļĪ£ ĒĢśļŖö ļŗ©ņØ╝ņ£ĀņĀäņ×Éļ│ĆņØ┤ļĪ£ ņØĖĒĢ£ ņ¦łĒÖśņ£╝ļĪ£, ļīĆļČĆļČäņØś ĒÖśņ×ÉļŖö ņä£ņä£Ē׳ ņ¦äĒ¢ēĒĢśļŖö ļ│┤Ē¢ēņןņĢĀļź╝ ņŻ╝ņåīļĪ£ ļ│æņøÉņŚÉ ļé┤ņøÉĒĢ£ļŗż. ļ│ĆņØ┤ ņ£ĀņĀäņ×ÉņØś ņóģļźśņŚÉ ļö░ļØ╝ ļ░£ļ│æņŗ£ĻĖ░Ļ░Ć ļŗżņ¢æĒĢśņŚ¼ ņśüņĢä, ņ£ĀņĢä, ņ▓ŁņåīļģäĻĖ░ņŚÉ ļ░£ļ│æĒĢśĻĖ░ļÅä ĒĢśĻ│Ā 30-40ļīĆ ņä▒ņØĖņØ┤ ļÉ£ ņØ┤ĒøäņŚÉ ņ▓½ ņ”ØņāüņØ┤ ļéśĒāĆļéśĻĖ░ļÅä ĒĢ£ļŗż. ĻĘĖ ņÖĖ ļÆżņä¼ņ£ĀĻĖ░ļæź ņ╣©ļ▓öņ£╝ļĪ£ ņØĖĒĢ£ ņ¦äļÅÖ ļ░Å ņ£äņ╣ś Ļ░ÉĻ░ü ņĀĆĒĢśņÖĆ ļ╣łļć©, ņĀłļ░Ģļć© ļō▒ ņåīļ│Ć ņ”ØņāüņØ┤ ĒØöĒĢśĻ▓ī ļÅÖļ░śļÉ£ļŗż. ņāüņ¦ĆņØś Ļ░Ģņ¦üņØĆ ņŚåņ£╝ļéś ņŗ¼ļČĆĻ▒┤ļ░śņé¼Ļ░Ć ĒĢŁņ¦äļÉśņ¢┤ ņ׳ļŖö Ļ▓ĮņÜ░Ļ░Ć ļ¦ÄĻ│Ā, Ļ▓ĮņÜ░ņŚÉ ļö░ļØ╝ ņĢäļלĒä▒ļ░śņé¼Ļ░Ć ĒĢŁņ¦äļÉśĻĖ░ļÅä ĒĢ£ļŗż. ņ×äņāü ņ¢æņāüņŚÉ ļö░ļØ╝ ņ¦äĒ¢ēņä▒ ĒĢśņ¦Ć Ļ░Ģņ¦üĻ│╝ ņØ╝ļČĆ ņåīļ│Ć ņ”Øņāü, ĒĢśņ¦Ć Ļ░ÉĻ░üņĀĆĒĢśļ¦īņØä ļ│┤ņØ┤ļŖö ņł£ņłśĒśĢ(pure form)Ļ│╝ ņØĖņ¦ĆĻĖ░ļŖźņĀĆĒĢś, ņŗ£ņŗĀĻ▓Įņ£äņČĢ, ņČöņ▓┤ņÖĖļĪ£ ņ”Øņāü, ņåīļćīņŗżņĪ░, ļ¦Éņ┤łņŗĀĻ▓Įļ│æ ļō▒ņØä ļÅÖļ░śĒĢ£ ļ│ĄĒĢ®ĒśĢ(complex form)ņ£╝ļĪ£ ĻĄ¼ļČäĒĢ£ļŗż[7]. ņØ┤ņĀäņŚÉļŖö pureņÖĆ complicatedļĪ£ ļČäļźśĒĢśĻĖ░ļÅä ĒĢśņśĆņ£╝ļéś, ņĄ£ĻĘ╝ņŚÉļŖö complex ņÜ®ņ¢┤ļź╝ ļŹö ļ¦ÄņØ┤ ņé¼ņÜ®ĒĢśĻ│Ā ņ׳ļŗż.
2. ņ¦äļŗ©
ņ£ĀņĀäĻ░Ģņ¦üĒĢśļ░śņŗĀļ¦łļ╣äņØś ĒÖĢņ¦äņØä ņ£äĒĢ┤ņä£ļŖö ņøÉņØĖ ņ£ĀņĀäņ×ÉņØś ļÅīņŚ░ļ│ĆņØ┤Ļ░Ć ĒÖĢņØĖļÉśņ¢┤ņĢ╝ ĒĢ£ļŗż. ĒĢśņ¦Ćļ¦ī ņÜ░ņäĀņĀüņ£╝ļĪ£ Ēøäņ▓£ ņ¦łĒÖśņ£╝ļĪ£ ņØĖĒĢ£ ņ¦äĒ¢ēņä▒ ĒĢśņ¦Ć Ļ░Ģņ¦üņØä ļ░░ņĀ£ĒĢśļŖö Ļ▓āņØ┤ ļ¼┤ņŚćļ│┤ļŗż ņżæņÜöĒĢśļŗż. Ļ░Éļ│äņØ┤ ĒĢäņÜöĒĢ£ ņŻ╝ņÜö ņ¦łĒÖśņØä Table 1ņŚÉ ņÜöņĢĮĒĢśņśĆļŗż. ņØ┤ļź╝Ēģīļ®┤ Ļ░Ćņן ĒØöĒĢ£ SPG4 ņ£ĀņĀäĻ░Ģņ¦üĒĢśļ░śņŗĀļ¦łļ╣äņØś ĒÅēĻĘĀ ļ░£ļ│æ ņŚ░ļĀ╣ņØĆ 31.7ņäĖņØ┤Ļ│Ā[6], ņä£ņä£Ē׳ ņ¦äĒ¢ēĒĢśļŖö Ļ▓ĮĻ│╝ļź╝ ļéśĒāĆļé┤ļŖö ļŹ░ ļ░śĒĢ┤, ņ×ÉĻ░Ćļ®┤ņŚŁ ņ¦łĒÖś ļśÉļŖö ļČĆņóģņ¢æņ”ØĒøäĻĄ░ņ£╝ļĪ£ ņØĖĒĢ£ ĒĢśņ¦ĆĻ░Ģņ¦üņØĆ ļīĆļČĆļČä ĻĖēņä▒ ļśÉļŖö ņĢäĻĖēņä▒ņ£╝ļĪ£ ļ░£ņāØĒĢśļ®░ ņøÉļ░£ņĖĪņéŁĻ▓ĮĒÖöņ”ØņØś Ļ▓ĮņÜ░ Ļ│ĀļĀ╣ņØś ļéśņØ┤ņŚÉ ļ░£ļ│æĒĢśĻ│Ā Ļ▒░ņØś ļ¬©ļōĀ Ļ▓ĮņÜ░ņŚÉ ĻĄ¼ļ¦łļ╣ä ņ”ØņāüņØ┤ ļéśĒāĆļé£ļŗż. ņĄ£ĻĘ╝ ļÅģļ”ĮņĀüņØĖ ņ¦łĒÖśņ£╝ļĪ£ ņĀĢļ”ĮļÉ£ MOG ĒĢŁņ▓┤ ņŚ░Ļ┤Ć ņ¦łĒÖśņØś Ļ▓ĮņÜ░, ņĢĮ 10% ĒÖśņ×ÉļŖö ņŗ¼ĒĢ£ ņŗĀĻ▓ĮĒĢÖņĀü ņ”ØņāüņŚÉļÅä ļČłĻĄ¼ĒĢśĻ│Ā ņ┤łĻĖ░ ļé┤ņøÉ ņŗ£ ņ▓Öņłś magnetic resonance imaging (MRI)ņŚÉņä£ ĒŖ╣ļ│äĒĢ£ ņØ┤ņāü ņåīĻ▓¼ņØä ļ│┤ņØ┤ņ¦Ć ņĢŖļŖö Ļ▓āņ£╝ļĪ£ ļéśĒāĆļé¼ļŗż[8]. ĻĘĖ ņÖĖ, ĒĢśņ¦Ć Ļ░Ģņ¦üņØ┤ ņä£ņä£Ē׳ ņĢģĒÖöļÉśņ¦Ćļ¦ī ļÜ£ļĀĘĒĢ£ MRI ņØ┤ņāüņØ┤ ļ░£Ļ▓¼ļÉśņ¦Ć ņĢŖļŖö ļŗżļźĖ ļŗ©ņØ╝ņ£ĀņĀäņ×É ņ¦łĒÖśņØä Ļ░Éļ│äĒĢ┤ņĢ╝ ĒĢ£ļŗż. ļ░▒ņ¦łņØ┤ņśüņ¢æņ”Ø, ļīĆņé¼ ņ¦łĒÖś, ņ▓ÖņłśņåīļćīņŗżņĪ░, Ļ░Ģņ¦üņŗżņĪ░ ļō▒ ļŗżņ¢æĒĢ£ ņ£ĀņĀä ņ¦łĒÖśņØ┤ ņŚ¼ĻĖ░ņŚÉ ĒżĒĢ©ļÉĀ ņłś ņ׳ļŖöļŹ░, ņØ┤ļōż ņ¦łĒÖśņØ┤ NGS ņ£ĀņĀäņ×É Ēī©ļäÉ Ļ▓Ćņé¼ņŚÉ ĒżĒĢ©ļÉśņ¦Ć ņĢŖņØä ņłś ņ׳ĻĖ░ ļĢīļ¼ĖņŚÉ ņŻ╝ņØśļź╝ ņÜöĒĢ£ļŗż. ĒŖ╣Ē׳ male-to-male transmissionņØ┤ ņŚåļŖö Ļ▓ĮņÜ░, ļČĆņŗĀļ░▒ņ¦łņØ┤ņśüņ¢æņ”Ø(adrenoleukodystrophy) Ļ░Éļ│äņØ┤ ĒĢäņÜöĒĢśļŗż(ABCD1) [9]. ĻĘĖ ņÖĖ ņ╣śļŻī ņśĄņģśņØ┤ ņ׳ļŖö ņ£ĀņĀä ņ¦łĒÖśņØĖ arginase deficiency, cerebrotendinous xanthomatosis, dopa-responsive dystonia, phenylketonuria, biotinidase deficiency, cobalamin-related remethylation disorders, methylenetetra hydrofolate reductase deficiency, primary coenzyme Q10 deficiencies ļō▒ņØś ņ¦łĒÖśņØä Ļ░Éļ│äĒĢ┤ņĢ╝ ĒĢ£ļŗż. ALS2, SPG11, SETX, VCP ņ£ĀņĀäņ×É ļ│ĆņØ┤ņØś Ļ▓ĮņÜ░, ĒÅēĻĘĀ ņāØņĪ┤ ĻĖ░Ļ░äņØ┤ ļ░£ļ│æ Ēøä 2-5ļģäņŚÉ ļČłĻ│╝ĒĢ£ ĻĘ╝ņ£äņČĢņä▒ņĖĪņéŁĻ▓ĮĒÖöņ”ØņØś ĒśĢĒā£ļĪ£ļÅä ļéśĒāĆļéĀ ņłś ņ׳ĻĖ░ ļĢīļ¼ĖņŚÉ ņ¦äļŗ©ņŚÉ Ļ░üļ│äĒĢ£ ņŻ╝ņØśļź╝ ņÜöĒĢ£ļŗż[10-16].
1) ņĀäĻĖ░ņāØļ”¼ĒĢÖņĀü Ļ▓Ćņé¼
ņ£ĀņĀäĻ░Ģņ¦üĒĢśļ░śņŗĀļ¦łļ╣ä ĒÖśņ×ÉņØś ņĀäĻĖ░ņāØļ”¼ĒĢÖņĀü Ļ▓Ćņé¼ņŚÉ ļīĆĒĢ£ ļé┤ņÜ®ņØĆ 2016ļģä ļ░£Ēæ£ļÉ£ ņØ┤Ēāłļ”¼ņĢä ņĮöĒśĖĒŖĖ ņŚ░ĻĄ¼ Ļ▓░Ļ│╝ļź╝ ņåīĻ░£ĒĢśĻ▓Āļŗż[17]. ņ▓┤ņä▒Ļ░ÉĻ░üņ£Āļ░£ņĀäņ£ä Ļ▓Ćņé¼(somatosensory evoked potential)ļź╝ ņŗ£Ē¢ēĒĢ£ ĒÖśņ×É 44ļ¬ģ ņżæ 30ļ¬ģņŚÉņä£ ņØ┤ņāüņØ┤ ĒÖĢņØĖļÉśņŚłļŗż(68.2%). ĒĢśņ¦ĆņÜ┤ļÅÖņ£Āļ░£ņĀäņ£ä Ļ▓Ćņé¼(motor evoked potential) Ļ▓░Ļ│╝, 32ļ¬ģ ņżæ 31ļ¬ģņŚÉņä£ ņżæņČöņÜ┤ļÅÖņĀäļÅäņŗ£Ļ░ä(central motor conduction time)ņØ┤ ņŚ░ņןļÉśņ¢┤ ņ׳Ļ▒░ļéś ņ£Āļ░£ļÉśņ¦Ć ņĢŖņĢśļŗż(96.9%). ņØ┤ļōż 31ļ¬ģ ņżæ 14ļ¬ģņØĆ ņāüņ¦ĆņÜ┤ļÅÖņ£Āļ░£ņĀäņ£ä Ļ▓Ćņé¼ņŚÉņä£ļÅä ņØ┤ņāüņØ┤ ĒÖĢņØĖļÉśņŚłļŗż(45.2%). ņŗĀĻ▓ĮņĀäļÅä Ļ▓Ćņé¼ ļ░Å ĻĘ╝ņĀäļÅä Ļ▓Ćņé¼ļź╝ ņŗ£Ē¢ēĒĢ£ 49ļ¬ģņØś ĒÖśņ×É ņżæ 23ļ¬ģņŚÉņä£ ņČĢņéŁĒśĢ ļ¦Éņ┤łņŗĀĻ▓Įļ│æņØ┤ ļÅÖļ░śļÉśņ¢┤ ņ׳ņŚłļŗż(46.9%). ĻĘĖ ņÖĖ 8ļ¬ģņØś SPG31 ĒÖśņ×Éļź╝ ļīĆņāüņ£╝ļĪ£ ĒĢ£ ļŹ┤ļ¦łĒü¼ ņŚ░ĻĄ¼ Ļ▓░Ļ│╝, 8ļ¬ģ ļ¬©ļæÉ ņŗĀĻ▓ĮĻĘ╝ņĀäļÅä Ļ▓Ćņé¼ņŚÉņä£ ļŗ©ņØ╝ņŗĀĻ▓Įļ│æņ”ØņØ┤ ĒÖĢņØĖļÉśņ¢┤ ņĢĢļ░Ģņ£╝ļĪ£ ņØĖĒĢ£ ņŗĀĻ▓ĮņåÉņāüņŚÉ ņĘ©ņĢĮĒĢ£ Ļ▓āņ£╝ļĪ£ ļ│┤ņØĖļŗż[18].
2) ņśüņāü Ļ▓Ćņé¼
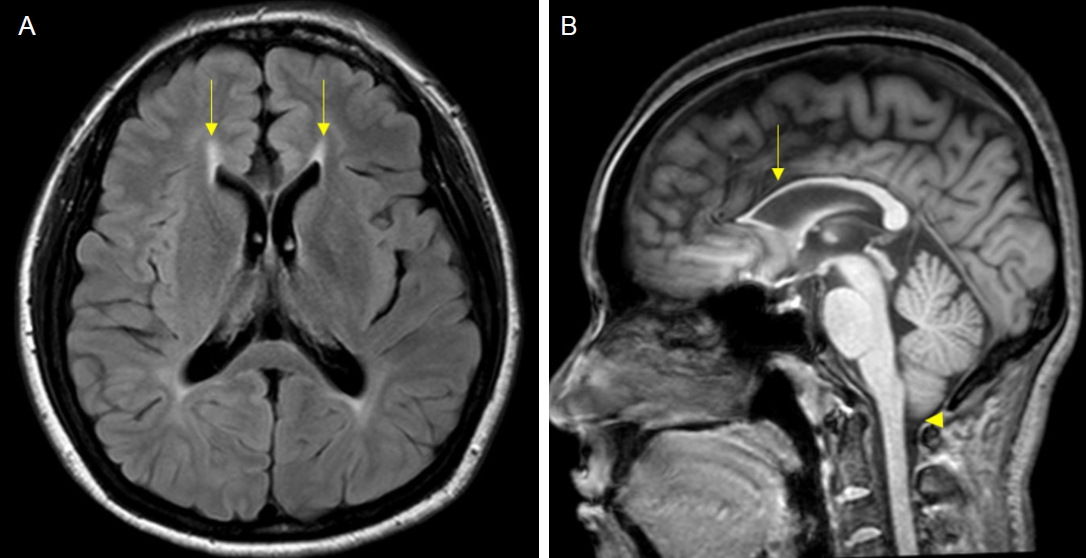
ļīĆļČĆļČäņØś ņ£ĀņĀäĻ░Ģņ¦üĒĢśļ░śņŗĀļ¦łļ╣ä ĒÖśņ×ÉļŖö ņ▓Öņłś MRIņŚÉņä£ ĒŖ╣ļ│äĒĢ£ ņØ┤ņāü ņåīĻ▓¼ņØä ļ│┤ņØ┤ņ¦Ć ņĢŖĻ│Ā, ņ▓ÖņłśņØś Ļ▓Įļ»ĖĒĢ£ ņ£äņČĢ ņĀĢļÅäļ¦ī Ļ┤Ćņ░░ļÉ£ļŗż[19]. ņ▓Öņłś ņŗżņ¦łļé┤ T2 ņŗĀĒśĖ ņ”ØĻ░Ć ļśÉļŖö T1 ņĪ░ņśü ņ”ØĻ░Ģ ļō▒ņØś ņØ┤ņāüņØ┤ ņ׳ļŖö Ļ▓ĮņÜ░ ļŗżļźĖ Ēøäņ▓£ ņ¦łĒÖśņØś Ļ░ĆļŖźņä▒ņØä ļŗżņŗ£ ĒĢ£ ļ▓ł Ļ│ĀļĀżĒĢ┤ļ│╝ Ļ▓āņØä ņČöņ▓£ĒĢ£ļŗż. ĻĘĖ ņÖĖ ļćī MRIĻ░Ć ņ£ĀņĀäĻ░Ģņ¦üĒĢśļ░śņŗĀļ¦łļ╣äņØś Ļ░Éļ│ä ņ¦äļŗ©ņŚÉ ļÅäņøĆņØä ņżä ņłś ņ׳ļŗż. ĒŖ╣Ē׳ SPG7ņØś Ļ▓ĮņÜ░ ņåīļćī ņ£äņČĢņØ┤ ļÅÖļ░śļÉśļŖö Ļ▓ĮņÜ░Ļ░Ć ĒØöĒĢśĻ│Ā ņåīļćīņ╣śņĢäĒĢĄ(dentate nucleus)ņØ┤ ĻĄÉļćī(pons)ņŚÉ ļ╣äĒĢ┤ T2 Ļ│ĀņŗĀĒśĖĻ░ĢļÅäļź╝ ļéśĒāĆļé╝ ņłś ņ׳ļŗż[20]. SPG11Ļ│╝ SPG15ņØś Ļ▓ĮņÜ░, ļćīļ¤ē ņ£äņČĢ(thinning of corpus callosum)Ļ│╝ ļćīņŗż ņŻ╝ņ£ä ļ░▒ņ¦łņØś T2 ņŗĀĒśĖ ņ”ØĻ░Ćļź╝ ļ│┤ņØ╝ ņłś ņ׳ļŗż(Fig. 2) [21,22]. SPG5ļź╝ ļ╣äļĪ»ĒĢ£ ņØ╝ļČĆ ņ£ĀņĀäĻ░Ģņ¦üĒĢśļ░śņŗĀļ¦łļ╣ä ĒÖśņ×ÉņŚÉņä£ ļ╣äĒŖ╣ņØ┤ņĀüņØĖ ļ░▒ņ¦ł ļ│æļ│ĆņØä ļ│┤ņØ╝ ņłś ņ׳ņ£╝ļéś[23], ņØ┤ļ¤¼ĒĢ£ Ļ▓ĮņÜ░ ļ░▒ņ¦łņØ┤ņśüņ¢æņ”Ø(leukodystrophy) Ļ░ĆļŖźņä▒ņØä ļ░░ņĀ£ĒĢ┤ņĢ╝ ĒĢ£ļŗż.
3) ņ£ĀņĀäņ×É Ļ▓Ćņé¼
ļ│┤Ļ│ĀņŚÉ ļö░ļØ╝ ņ░©ņØ┤Ļ░Ć ņ׳ņ£╝ļéś ņ£ĀņĀäĻ░Ģņ¦üĒĢśļ░śņŗĀļ¦łļ╣ä ĒÖśņ×ÉņØś 13-40%ņŚÉņä£ Ļ░ĆņĪ▒ļĀźņØ┤ ņŚåļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż[3,4]. NGS ļ╣äņÜ®ņØ┤ Ļ░ÉņåīĒĢ©ņŚÉ ļö░ļØ╝ NGS ĻĖ░ļ░ś ņ£ĀņĀäņ×É Ēī©ļäÉ Ļ▓Ćņé¼Ļ░Ć ļ│┤ĒÄĖĒÖöļÉśĻ│Ā ņ׳ņ£╝ļéś, ņØ┤ļ¤¼ĒĢ£ Ļ▓Ćņé¼ Ļ▓░Ļ│╝ļź╝ ĒĢ┤ņäØĒĢśĻĖ░ ņ£äĒĢ┤ņä£ļŖö NGS ĻĖ░ņłĀņØś ĒĢ£Ļ│äņŚÉ ļīĆĒĢ£ ņØ┤ĒĢ┤Ļ░Ć ĒĢäņÜöĒĢśļŗż. ņ▓½ņ¦Ė, NGS Ļ▓Ćņé¼ļŖö Ļ▒░ļīĆ Ļ▓░ņŗż(large deletion) ļśÉļŖö ņżæļ│Ą(duplication)ņØä ĒżĒĢ©ĒĢ£ ņ£ĀņĀäņ×É ļ│ĄņĀ£ņłś ļ│ĆņØ┤(copy number variation)ļź╝ Ļ▓ĆņČ£ĒĢśĻĖ░ ņ¢┤ļĀĄļŗż. ļæśņ¦Ė, ĒöäļĪ£ļ¬©Ēä░(promoter) ļśÉļŖö ņØĖĒŖĖļĪĀ ņĢłņ¬Į(deep intron)ņŚÉ ņ£äņ╣śĒĢ£ ļ│ĆņØ┤ļź╝ ņ░ŠņĢäļé┤ĻĖ░ ņ¢┤ļĀĄļŗż. ļ¦łņ¦Ćļ¦ēņ£╝ļĪ£ ņé╝ĒĢĄņé░ ļ░śļ│Ąņä£ņŚ┤ ņ¦łĒÖś(triple repeat disorders)ņØä ņ¦äļŗ©ĒĢśĻĖ░ ņ¢┤ļĀĄļŗż[24]. ĒŖ╣Ē׳ SPAST ļ│ĆņØ┤ļĪ£ ņØĖĒĢ£ SPG4ņØś Ļ▓ĮņÜ░, ņŚæņåÉ Ļ▓░ņŗżņØ┤ ņāüļīĆņĀüņ£╝ļĪ£ ĒØöĒĢśĻĖ░ ļĢīļ¼ĖņŚÉ NGS ņ£ĀņĀäņ×É Ēī©ļäÉ Ļ▓Ćņé¼ņŚÉņä£ ļ│æņĀü ļ│ĆņØ┤Ļ░Ć Ļ▓ĆņČ£ļÉśņ¦Ć ņĢŖņĢśļŹöļØ╝ļÅä ņ£ĀņĀäĻ░Ģņ¦üĒĢśļ░śņŗĀļ¦łļ╣äņØś Ļ░ĆņĪ▒ļĀźņØ┤ ņ׳Ļ▒░ļéś ņ×äņāüņĀüņ£╝ļĪ£ ņØśņŗ¼ļÉśļŖö Ļ▓ĮņÜ░ multiplex ligation-dependent probe amplification Ļ▓Ćņé¼ļź╝ ņŗ£Ē¢ēĒĢśļŖö Ļ▓āņØä ņČöņ▓£ĒĢ£ļŗż.
3. ņāüņŚ╝ņāēņ▓┤ ņÜ░ņä▒ ņ£ĀņĀä(Table 2)
1) SPG4
ņĀäņ▓┤ ņ£ĀņĀäĻ░Ģņ¦üĒĢśļ░śņŗĀļ¦łļ╣ä ņżæ Ļ░Ćņן ĒØöĒĢ£ ĒśĢĒā£ļĪ£, SPAST ņ£ĀņĀäņ×É ļ│ĆņØ┤ņŚÉ ņØśĒĢ┤ ļ░£ņāØĒĢ£ļŗż[25]. SPASTļŖö ļ»ĖņäĖņåīĻ┤ĆņØä ņĀłļŗ©ĒĢśĻ│Ā ņ×¼ņä▒ņןņØä ņ┤ēņ¦äĒĢśļŖö Spastin ļŗ©ļ░▒ņ¦łņØä ņĢöĒśĖĒÖöĒĢ£ļŗż. 608ļ¬ģņØś ĒÖśņ×Éļź╝ ļīĆņāüņ£╝ļĪ£ ĒĢ£ ļīĆĻĘ£ļ¬© ņŚ░ĻĄ¼ņŚÉņä£ ņāüņŚ╝ņāēņ▓┤ ņÜ░ņä▒ ļ░Å ņŚ┤ņä▒ ņ£ĀņĀäņØä ĒżĒĢ©ĒĢ£ ņĀäņ▓┤ ņ£ĀņĀäĻ░Ģņ¦üĒĢśļ░śņŗĀļ¦łļ╣ä ĒÖśņ×ÉņØś 1/3ņØ┤ SPG4ņØĖ Ļ▓āņ£╝ļĪ£ ļ│┤Ļ│ĀļÉśņŚłņ£╝ļ®░, ņāüņŚ╝ņāēņ▓┤ ņÜ░ņä▒ ņ£ĀņĀä Ļ░ĆņĪ▒ļĀźņØ┤ ņ׳ļŖö Ļ▓ĮņÜ░ 60%, Ļ░ĆņĪ▒ļĀźņØ┤ ņŚåļŖö ņé░ļ░£ņä▒ņØś Ļ▓ĮņÜ░ 15%ļź╝ ņ░©ņ¦ĆĒĢśņśĆļŗż[3]. ĒÅēĻĘĀ ļ░£ļ│æ ņŚ░ļĀ╣ņØĆ 31.7ņäĖņØ┤ļéś 70ņäĖņŚÉ ļ░£ļ│æĒĢ£ ĒÖśņ×ÉņŚÉ ļīĆĒĢ£ ļ│┤Ļ│ĀļÅä ņ׳ļŗż[6]. ļīĆļČĆļČäņØś Ļ▓ĮņÜ░ ļ░£ļ│æ ņ┤łĻĖ░ņŚÉļŖö ĒĢśņ¦Ć Ļ░Ģņ¦ü ļŗ©ļÅģņ£╝ļĪ£ ļéśĒāĆļéśļŖö ņł£ņłśĒśĢņØ┤ļŗż[6]. 196ļ¬ģņØä ļīĆņāüņ£╝ļĪ£ ĒĢ£ ļÅģņØ╝ ņĮöĒśĖĒŖĖ ņŚ░ĻĄ¼ Ļ▓░Ļ│╝ņŚÉ ļö░ļź┤ļ®┤ ņĢĮ 5-10%ņŚÉņä£ ņŗżņĪ░ ļśÉļŖö ļ¦Éņ┤ł ņÜ┤ļÅÖņŗĀĻ▓Į ņ╣©ļ▓öņØ┤ ļÅÖļ░śļÉśņŚłĻ│Ā, ļō£ļ¼╝Ļ▓ī ņØĖņ¦ĆĻĖ░ļŖźņĀĆĒĢś, ĻĄ¼ļ¦łļ╣ä, ņČöņ▓┤ņÖĖļĪ£ ņ”ØņāüņØ┤ ļ│┤Ļ│ĀļÉśĻĖ░ļÅä ĒĢśņśĆļŗż[3].
2) SPG3A
ļæÉ ļ▓łņ¦ĖļĪ£ ĒØöĒĢ£ ņāüņŚ╝ņāēņ▓┤ ņÜ░ņä▒ ņ£ĀņĀäĻ░Ģņ¦üĒĢśļ░śņŗĀļ¦łļ╣äļĪ£, Atlastin ļŗ©ļ░▒ņ¦łņØä ņĢöĒśĖĒÖöĒĢśļŖö ATL1 ņ£ĀņĀäņ×É ļ│ĆņØ┤ņŚÉ ņØśĒĢ┤ ļ░£ņāØĒĢ£ļŗż. AtlastinņØĆ ņåīĒżņ▓┤(endoplasmic reticulum) ļ¦ēņŚÉ ņĪ┤ņ×¼ĒĢśļŖö GTPaseļĪ£ ņåīĒżņ▓┤ ļ¦ēņØś homotypic fusionņØä ļ¦żĻ░£ĒĢśļŖö ņŚŁĒĢĀņØä ĒĢ£ļŗż. SPG3AņØś ņ×äņāü ņ¢æņāüņØĆ SPG4ņÖĆ Ļ▒░ņØś ņ£Āņé¼ĒĢśļéś ņČĢņéŁĒśĢ ļ¦Éņ┤łņŗĀĻ▓Įļ│æņØ┤ ņĪ░ĻĖł ļŹö ĒØöĒĢśĻ▓ī ļ│┤Ļ│ĀļÉ£ļŗż(25%) [3]. ļ░£ļ│æ ņŚ░ļĀ╣ņØ┤ SPG4ņŚÉ ļ╣äĒĢ┤ Ēø©ņö¼ ņ¢┤ļ”¼ļŗż(ĒÅēĻĘĀ 5.6ņäĖ) [3].
3) SPG31
ņäĖ ļ▓łņ¦ĖļĪ£ ĒØöĒĢ£ ņāüņŚ╝ņāēņ▓┤ ņÜ░ņä▒ ņ£ĀņĀäĻ░Ģņ¦üĒĢśļ░śņŗĀļ¦łļ╣äļĪ£, ļ»ĖĒåĀņĮśļō£ļ”¼ņĢä ļŗ©ļ░▒ņ¦łņØĖ receptor expression-enhancing protein 1ņØä ņĢöĒśĖĒÖöĒĢśļŖö REEP1 ņ£ĀņĀäņ×É ļ│ĆņØ┤ņŚÉ ņØśĒĢ┤ ļ░£ņāØĒĢ£ļŗż[26]. ņ×äņāü ņ¢æņāüņØĆ SPG4 ļ░Å SPG3AņÖĆ ņ£Āņé¼ĒĢśļéś, ņĢĮ 50%ņŚÉņä£ ņČĢņéŁĒśĢ ļ¦Éņ┤łņŗĀĻ▓Įļ│æņØ┤ ļÅÖļ░śļÉ£ļŗż[27]. ļ░£ļ│æ ņŚ░ļĀ╣ņØĆ 10ļīĆņÖĆ 40ļīĆĻ░Ć ļ¦Äņ£╝ļéś(bimodal distribution), Ļ░ĆĻ│äņŚÉ ļö░ļØ╝ ļ¦żņÜ░ ļŗżņ¢æĒĢ£ Ļ▓āņ£╝ļĪ£ ļ│┤Ļ│ĀļÉśĻ│Ā ņ׳ļŗż[27].
4) SPG30
ļ»ĖņäĖņåīĻ┤ĆņŚÉ Ļ▓░ĒĢ®ĒĢśņŚ¼ ņåīĒż(vesicle) ļ░Å ņäĖĒżņåīĻĖ░Ļ┤ĆņØä ņÜ┤ļ░śĒĢśļŖö ņŚŁĒĢĀņØä ĒĢśļŖö Kinesin-like protein KIF1A ļŗ©ļ░▒ņ¦łņØä ņĢöĒśĖĒÖöĒĢśļŖö KIF1A ņ£ĀņĀäņ×É ļ│ĆņØ┤ņŚÉ ņØśĒĢ┤ ļ░£ņāØĒĢ£ļŗż. ņāüņŚ╝ņāēņ▓┤ ņÜ░ņä▒ ņ£ĀņĀäņ£╝ļĪ£ ļČäļźśļÉśņŚłņ£╝ļéś, ļō£ļ¼╝Ļ▓ī ņāüņŚ╝ņāēņ▓┤ ņŚ┤ņä▒ņ£╝ļĪ£ ņ£ĀņĀäļÉśĻĖ░ļÅä ĒĢ£ļŗż[28,29]. ņÜ░ņä▒ ņ£ĀņĀäņØś Ļ▓ĮņÜ░ ļ░£ļ│æ ņŚ░ļĀ╣ņØ┤ 0-57ņäĖļĪ£ ļ¦żņÜ░ ļŗżņ¢æĒĢśļéś 50% ņØ┤ņāüņØś ĒÖśņ×ÉļŖö 11ņäĖ ņØ┤ņĀäņŚÉ ņ▓½ ņ”ØņāüņØ┤ ļ░£ņāØĒĢśņśĆļŗż[28]. 30ļ¬ģ ņżæ 11ļ¬ģņŚÉņä£ ļÆżņä¼ņ£ĀĻĖ░ļæź ņ╣©ļ▓öņØ┤ ņ׳ņŚłĻ│Ā, 13ļ¬ģņŚÉņä£ ĒĢśņ¦Ć ĻĘ╝ņ£äļČĆ ĻĘ╝ļĀźņĀĆĒĢś ļ░Å ĻĘ╝ņ£äņČĢņØ┤ ĒÖĢņØĖļÉśņ¢┤ ņČĢņéŁĒśĢ ļ¦Éņ┤łņŗĀĻ▓Įļ│æņØ┤ ļÅÖļ░śļÉśņŚłņØä Ļ▓āņ£╝ļĪ£ ņČöņĀĢļÉ£ļŗż. ļō£ļ¼╝Ļ▓ī ņØĖņ¦ĆĻĖ░ļŖźņĀĆĒĢśĻ░Ć ļÅÖļ░śļÉĀ ņłś ņ׳ļŗż[28].
4. ņāüņŚ╝ņāēņ▓┤ ņŚ┤ņä▒ ņ£ĀņĀä(Table 3)
1) SPG11
ņāüņŚ╝ņāēņ▓┤ ņŚ┤ņä▒ ņ£ĀņĀäĻ░Ģņ¦üĒĢśļ░śņŗĀļ¦łļ╣ä ņżæ Ļ░Ćņן ĒØöĒĢ£ ĒśĢĒā£ļĪ£, Spatacsin ļŗ©ļ░▒ņ¦łņØä ņĢöĒśĖĒÖöĒĢśļŖö SPG11 ņ£ĀņĀäņ×É ļ│ĆņØ┤ņŚÉ ņØśĒĢ┤ ļ░£ņāØĒĢ£ļŗż. ļ░£ļ│æ ņŚ░ļĀ╣ņØĆ 4-36ņäĖļĪ£ ļ│┤Ļ│ĀļÉśĻ│Ā ņ׳ņ£╝ļ®░ ņ×äņāü ņ¢æņāüņØ┤ ļ¦żņÜ░ ļŗżņ¢æĒĢśļŗż. ļīĆļČĆļČäņØś ĒÖśņ×ÉņŚÉņä£ ņØĖņ¦ĆĻĖ░ļŖźņĀĆĒĢśĻ░Ć ļÅÖļ░śļÉśĻ│Ā ņŗ¼ņ¦Ćņ¢┤ ĒĢśņ¦Ć Ļ░Ģņ¦üļ│┤ļŗż ļ©╝ņĀĆ ļéśĒāĆļéśļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż[21]. ņĢĮ 50% ĒÖśņ×ÉņŚÉņä£ ņČĢņéŁĒśĢ ļ¦Éņ┤łņŗĀĻ▓Įļ│æ, ņŗżņĪ░, ĻĄ¼ļ¦łļ╣ä ļō▒ņØś ņ”ØņāüņØ┤ ļÅÖļ░śļÉ£ļŗż[21]. ņāüņ¦Ć Ļ░Ģņ¦üņ£╝ļĪ£ ņ¦äĒ¢ēļÉśļŖö Ļ▓ĮņÜ░Ļ░Ć ļ¦ÄĻ│Ā, ņČöņ▓┤ņÖĖļĪ£ ņ”Øņāü, Ļ▓ĮļĀ©, ņŗ£ņŗĀĻ▓Į ņ£äņČĢ ļō▒ņØ┤ ļÅÖļ░śļÉ£ ņé¼ļĪĆļÅä ļ│┤Ļ│ĀļÉśņŚłļŗż[5].
2) SPG5
SPG5ļŖö cytochrome P-450 oxysterol 7a-hydroxylaseļź╝ ņĢöĒśĖĒÖöĒĢśļŖö CYP7B1 ņ£ĀņĀäņ×É ļ│ĆņØ┤ņŚÉ ņØśĒĢ┤ ļ░£ņāØĒĢ£ļŗż[30]. 25-hydroxycholesterolĻ│╝ 27-hydroxycholesterol (27-OHC)ņØĆ CYP7B1ņŚÉ ņØśĒĢ┤ņä£ 7a-hydroxylation ļÉśļŖöļŹ░, CYP7B1 ļ│ĆņØ┤ņŚÉ ņØśĒĢ┤ ņČĢņĀüļÉśļŖö 27-OHCĻ░Ć ņŗĀĻ▓ĮĒć┤Ē¢ēņä▒ ļ│ĆĒÖöļź╝ ņØ╝ņ£╝ĒéżļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż[31]. SPG5ļŖö ņāüļīĆņĀüņ£╝ļĪ£ ļō£ļ¼Ė Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĪīņ£╝ļéś, ņżæĻĄŁ ņĮöĒśĖĒŖĖņŚÉņä£ļŖö 101ļ¬ģņØś ņāüņŚ╝ņāēņ▓┤ ņŚ┤ņä▒ ņ£ĀņĀä ņ╝ĆņØ┤ņŖż ņżæ 28ļ¬ģņØ┤ SPG5ļĪ£ ļ│┤Ļ│ĀļÉśņŚłļŗż[3,4,23,32]. 34ļ¬ģņØś ĒÖśņ×Éļź╝ ļīĆņāüņ£╝ļĪ£ ĒĢ£ ņ£Āļ¤Į ļŗżĻĖ░Ļ┤Ć ņŚ░ĻĄ¼ņŚÉņä£ ĒÅēĻĘĀ ļ░£ļ│æ ņŚ░ļĀ╣ņØĆ 13ņäĖņśĆĻ│Ā ņĢĮ 94%ņØś ĒÖśņ×ÉņŚÉņä£ ļÆżņä¼ņ£ĀĻĖ░ļæź ņ╣©ļ▓öņ£╝ļĪ£ ņØĖĒĢ£ Ļ░ÉĻ░ü ņĀĆĒĢśĻ░Ć ņ׳ņŚłņ£╝ļ®░ 47%ļŖö Ļ░ÉĻ░üņŗżņĪ░ļź╝ ļÅÖļ░śĒĢśņśĆļŗż. ņØĖņ¦ĆĻĖ░ļŖź ņĀĆĒĢśļŖö ļ│┤Ļ│ĀļÉśņ¦Ć ņĢŖņĢśļŗż[33].
3) SPG7
Paraplegin ļŗ©ļ░▒ņ¦łņØä ņĢöĒśĖĒÖöĒĢśļŖö SPG7 ņ£ĀņĀäņ×É ļ│ĆņØ┤ņŚÉ ņØśĒĢ┤ ļ░£ņāØĒĢ£ļŗż. ļ░£ļ│æ ņŚ░ļĀ╣ņØ┤ ĒÅēĻĘĀ 41.7ņäĖļĪ£ ļŖ”ņØĆ ĒÄĖņØ┤ļ®░ ņŚ¼ņ×Éļ│┤ļŗż ļé©ņ×ÉĻ░Ć ņĪ░ĻĖł ļŹö ļ¦Äļŗż[3,20,34]. SPG11Ļ│╝ ļ¦łņ░¼Ļ░Ćņ¦ĆļĪ£ ļ│ĄĒĢ®ĒśĢņØ┤ ļīĆļČĆļČäņØ┤Ļ│Ā, 50% ņØ┤ņāüņØś ĒÖśņ×ÉņŚÉņä£ ņåīļćīņŗżņĪ░ ļō▒ ņåīļćī ņ¦ĢĒøäĻ░Ć Ļ┤Ćņ░░ļÉśņ¢┤ MRI ņØ┤ņāü ņåīĻ▓¼ņŚÉ ļČĆĒĢ®ĒĢ£ļŗż[20,35,36]. ņāüĒĢśņ¦Ć ĻĘ╝ņ£äļČĆ ĻĘ╝ļĀźņĀĆĒĢśņÖĆ ļ¦īņä▒ņ¦äĒ¢ēņÖĖņĢłĻĘ╝ļ¦łļ╣äĻ░Ć ņāüļīĆņĀüņ£╝ļĪ£ ĒØöĒĢśĻ▓ī ļéśĒāĆļéśļ®░, ļō£ļ¼╝Ļ▓ī ņŗ£ņŗĀĻ▓Įļ│æņ”ØņØ┤ ļÅÖļ░śļÉśĻĖ░ļÅä ĒĢ£ļŗż[37].
4) SPG15
Spastizin ļŗ©ļ░▒ņ¦łņØä ņĢöĒśĖĒÖöĒĢśļŖö ZFYVE26 ņ£ĀņĀäņ×É ļ│ĆņØ┤ņŚÉ ņØśĒĢ┤ ļ░£ņāØĒĢ£ļŗż. ņ×äņāü ņ¢æņāüņØĆ SPG11Ļ│╝ ņ£Āņé¼ĒĢśļéś ņČĢņéŁĒśĢ ļ¦Éņ┤łņŗĀĻ▓Įļ│æĻ│╝ ĒīīĒé©ņŖ© ņ”ØņāüņØ┤ ņĪ░ĻĖł ļŹö ĒØöĒĢśĻ▓ī ļÅÖļ░śļÉ£ļŗż[21].
Ļ▓░ ļĪĀ
ņØ┤ņāüņ£╝ļĪ£ ņ£ĀņĀäĻ░Ģņ¦üĒĢśļ░śņŗĀļ¦łļ╣äņØś ņ×äņāü ņ¢æņāüĻ│╝ ņ£ĀņĀäņ×É Ļ▓Ćņé¼ļź╝ ĒåĄĒĢ£ ņ¦äļŗ© Ļ│╝ņĀĢņŚÉ ļīĆĒĢ┤ ņĢīņĢäļ│┤ņĢśļŗż. ņ£ĀņĀäĻ░Ģņ¦üĒĢśļ░śņŗĀļ¦łļ╣äņØś ņ¦äļŗ©ņØä ņ£äĒĢ┤ņä£ļŖö ņ╣śļŻī ļ░®ļ▓ĢņØ┤ ņ׳ļŖö Ēøäņ▓£ ņ¦łĒÖśĻ│╝ ņØ╝ļČĆ ņ£ĀņĀä ņ¦łĒÖśņØä Ļ░Éļ│äĒĢśļŖö Ļ▓āņØ┤ ņżæņÜöĒĢśļŗż. ņĄ£ĻĘ╝ NGS ņ£ĀņĀäņ×É Ēī©ļäÉ Ļ▓Ćņé¼Ļ░Ć ņĀÉņ░© ĒÖ£ņä▒ĒÖöļÉśĻ│Ā ņ׳ņ£╝ļéś, Ļ░Ćņן ĒØöĒĢ£ SPG4 ņøÉņØĖ ņ£ĀņĀäņ×ÉņØĖ SPAST ņŚæņåÉ Ļ▓░ņŗżĻ│╝ Ļ░ÖņØĆ ļ│ĆņØ┤ļŖö NGS Ļ▓Ćņé¼ņŚÉņä£ ļ░£Ļ▓¼ĒĢśĻĖ░ ņ¢┤ļĀĄĻ│Ā, ĻĘĖ ņÖĖ ļÜ£ļĀĘĒĢ£ MRI ņØ┤ņāüņØ┤ ļÅÖļ░śļÉśņ¦Ć ņĢŖļŖö ļŗżļźĖ ņ£ĀņĀä ņ¦łĒÖś ļśÉĒĢ£ Ļ░Éļ│äĒĢ┤ņĢ╝ ĒĢśĻĖ░ ļĢīļ¼ĖņŚÉ ņ£ĀņĀäĻ░Ģņ¦üĒĢśļ░śņŗĀļ¦łļ╣äņØś ņ£ĀņĀäĒśĢ-Ēæ£ĒśäĒśĢ ņāüĻ┤ĆĻ┤ĆĻ│äļź╝ ņĀĢĒÖĢĒ׳ ņØ┤ĒĢ┤ĒĢśļŖö Ļ▓āņØ┤ ņżæņÜöĒĢśļŗż.